
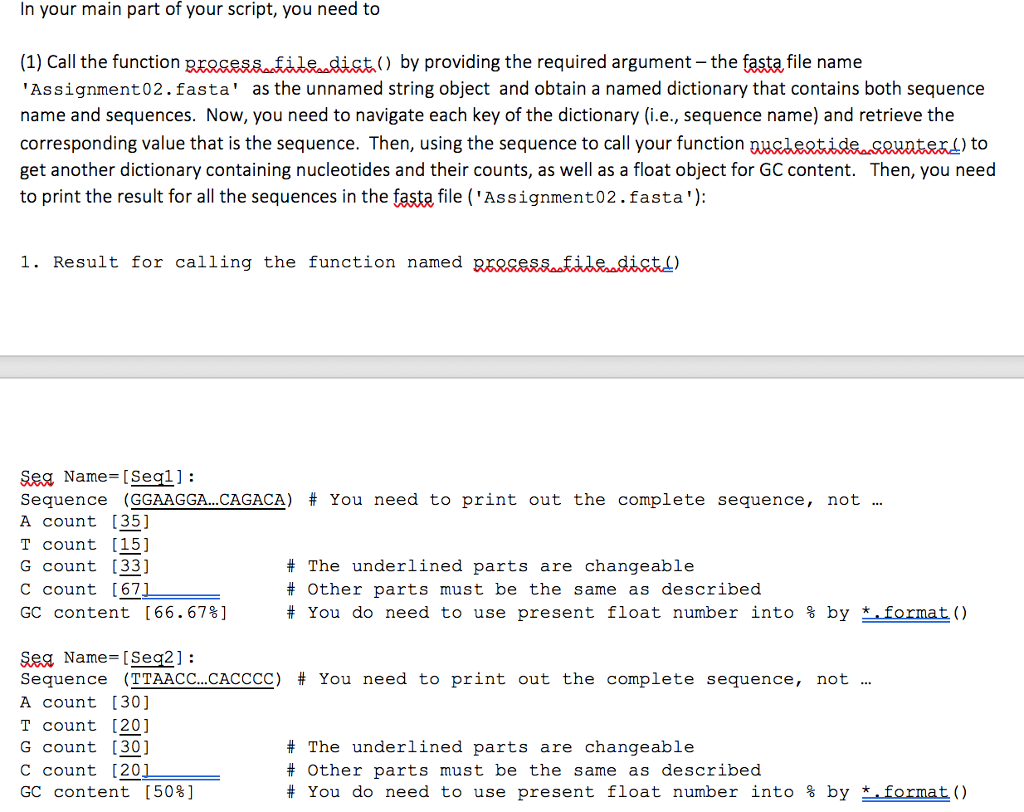

def Rrosesstile lisitienamg): # Given a file name in string object, you need to open this file and save sequence # names and sequences as a tuple into an empty list. # Then, your function will return a list # The returned dictionary is named as saist (eBr [( ' seq1 ' , ' GGAGGA return seg Jist 'L-) In your main part of your script, you need to (1) Call the function process fildist() by providing the required argument - the fasta file name 'Assignment02.fasta' as the unnamed string object and obtain a named dictionary that contains both sequence name and sequences. Now, you need to navigate each key of the dictionary (i.e., sequence name) and retrieve the corresponding value that is the sequence. Then, using the sequence to call your function get another dictionary containing nucleotides and their counts, as well as a float object for GC content. Then, you need to print the result for all the sequences in the fast file ('Assignment02.fasta'): ) to 1. Result for calling the function named RESSesaatabe. tt) SRA Name= [ Seal ] : Seguence (GGAAGGA CAGACA) # You need to print out the complete seguence, not A count [35] T count 15] G count 33] c count [67 GC content [66.67%] # The underlined parts are changeable # Other parts must be the same as described # You do need to use present float number into % by *. format() SRA Name= [Sea2] : Sequence (TTAACC CACCCC) # You need to print out the complete sequence, not A count [30] T count [20] G count [30] C count [201 GC content [50%] # The underlined parts are changeable # Other parts must be the same as described # You do need to use present float number into % by (1) Call the function process file list) by providing the required argumentthe fasta, file name Assignment02.fasta' as the unnamed string object and obtain a named list that contains both sequence name and sequences as the two elements within a tuple. Now, you need to navigate each element of your list to retrieve both the sequence name and sequence. Then, using the sequence to call your function dictionary containing nucleotides and their counts, as well as a float object for GC content. Then, you need to print the result for all the sequences in the fast file ('Assignment02.fasta'): to get a 2. Result for calling the function named eroceS file ist.) Sea Name- [Seql1: Seguence (GGAAGGA CAGACA) # You need to print out the complete sequence, not A count [351 T count [151 G count [33] C count [67 GC content [66.67%] # The underlined parts are changeable # Other parts must be the same as described # You do need to use present float number into % by- Sea Name [Seq2]: Seguence (TTAACC.. CAC CCC) # You need to print out the complete seguence, not A count [30] T count [20] G count [30] c count [20 GC content [50%] # The underlined parts are changeable # Other parts must be the same as described # You do need to use present float number into % byformat () - # Show results for Sea3 and SeaA Make sure that the actually results of 1 and 2 should be the same




