

Question: please try to answer parts 3,7&8 especially using python 2.7. my code is not working and this is due today. verify using working test codes.
please try to answer parts 3,7&8 especially using python 2.7. my code is not working and this is due today. verify using working test codes. 





Basically I am stuck with the longestORF non reading (part 3 of the assignment)
the testcode for that is a file called X73525 found in this NCBI file: Next, you should download the salmonella sequence we discussed in the text, accession number X73525, from NCBI as a fasta file. Go here for a reminder of how to do this. The file should be called X73525.fa (rename it if it's not). Move it to the same directory as your geneFinder.py file.
#testcode X73525=loadSeq("X73525.fasta") print "The longest ORF noncoding for the fasta file (X73525.fasta)with 50 numReps is:", longestORFNoncoding(X73525,50) This should return 623 or somewhere in that range. Thank you!!
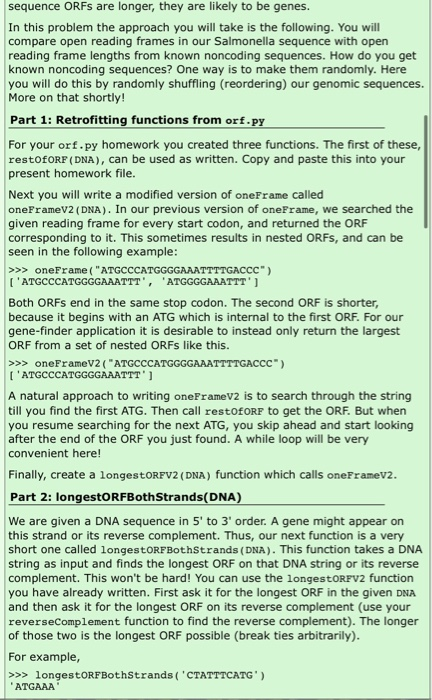
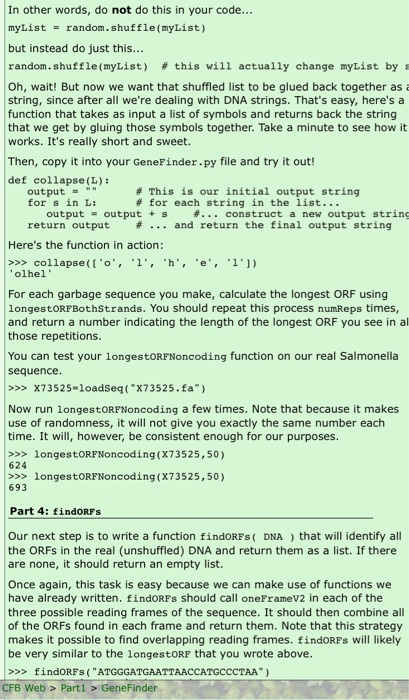
Gene finder All of your code for this problem should be in a file called GeneFinder.py. At the end you will also make a file with comments called GeneFinder.txt. Introduction In the text we discussed some investigations of Salmonella pathogenisis. Researchers identified genomic regions present in Salmonella but not in closely related species such as E. coli. Further study revealed that some of these regions are essential to Salmonella-specific pathogenic processes. One particular region was found to be needed for the entry of the bacterium into the epithelial cells of the gut. The researchers studying it wished to identify the protein-coding genes in it, so they sequenced it and analyzed that sequence for genes. In this homework, you will develop a simple gene finder, and use it to examine this sequence. You will also examine a related "mystery" sequence, and try to determine its function. You can begin by creating a homework file called GeneFinder.py. You will make use of a number of functions we have already written. You can give yourself access to the functions in your load.py and dna.py files by making sure they are present in the same directory as GeneFinder.py. Then include this at the top of GeneFinder.py: from load import * from dna import * Next, you should download the salmonella sequence we discussed in the text, accession number X73525, from NCBI as a fasta file. Go here for a reminder of how to do this. The file should be called x73525. fa (rename it if it's not). Move it to the same directory as your geneFinder.py file. A simple gene-finding strategy A simple gene-finding strategy is to look for large open reading frames. As we've seen, an open reading frame (ORF) is the stretch of sequence between a start codon and the next in frame stop codon. This strategy depends on the assumption that protein-coding sequence has larger ORFS than noncoding sequence. In noncoding regions "Start" and "Stop" codons appear by random chance, and the ORFs between them are not long. In many circumstances protein-coding genes have ORFs that are longer than this. To look for genes in a particular sequence, you can identify its longest ORFs. But how do you know if these are genes? To help decide, you can look at many known noncoding regions to obtain a distribution of open reading frame lengths for noncoding DNA. You can then compare the longest ORFs in our test sequence with this distribution. If the test sequence ORFs are longer, they are likely to be genes. In this problem the approach you will take is the following. You will compare open reading frames in our Salmonella sequence with open sequence ORFs are longer, they are likely to be genes. In this problem the approach you will take is the following. You will compare open reading frames in our Salmonella sequence with open reading frame lengths from known noncoding sequences. How do you get known noncoding sequences? One way is to make them randomly. Here you will do this by randomly shuffling (reordering) our genomic sequences More on that shortly! Part 1: Retrofitting functions from orf.py For your orf.py homework you created three functions. The first of these, restOfORP (DNA), can be used as written. Copy and paste this into your present homework file. Next you will write a modified version of oneFrame called oneFrameV2 (DNA). In our previous version of oneFrame, we searched the given reading frame for every start codon, and returned the ORF corresponding to it. This sometimes results in nested ORFs, and can be seen in the following example: >>> oneFrame("ATGCCCATGGGGAAATTTTGACCC") ['ATGCCCATGGGGAAATTT', 'ATGGGGAAATTT') Both ORFs end in the same stop codon. The second ORF is shorter, because it begins with an ATG which is internal to the first ORF. For our gene-finder application it is desirable to instead only return the largest ORF from a set of nested ORFs like this. >>> oneFrameV2 ("ATGCCCATGGGGAAATTTTGACCC") ('ATGCCCATGGGGAAATTT' ) A natural approach to writing oneFrameV2 is to search through the string till you find the first ATG. Then call restOFORF to get the ORF. But when you resume searching for the next ATG, you skip ahead and start looking after the end of the ORF you just found. A while loop will be very convenient here! Finally, create a longestORFV2 (DNA) function which calls oneFramev2. Part 2: longestORFBoth Strands(DNA) We are given a DNA sequence in 5' to 3' order. A gene might appear on this strand or its reverse complement. Thus, our next function is a very short one called longestORFBothStrands (DNA). This function takes a DNA string as input and finds the longest ORF on that DNA string or its reverse complement. This won't be hard! You can use the longestORFV2 function you have already written. First ask it for the longest ORF in the given DNA and then ask it for the longest ORF on its reverse complement (use your reverseComplement function to find the reverse complement). The longer of those two is the longest ORF possible (break ties arbitrarily). For example, >>> longestORFBothStrands('CTATTTCATG') ATGAAA Pause here to make sure you understand how we got that answer. Part 3: longestORFNoncoding (DNA, numReps) To assess whether long ORFs are genes, a researcher would ask the question, "is this sequence length indicative of a coding region or would I expect to see sequences this long in garbage?" By "garbage", of course, we mean just random sequences of nucleotides. We'll test this by generating a bunch of "garbage" sequences of the same length as our test DNA sequence, and measuring the maximum ORF length in each. Then, we'll ask the following question. Is the very longest ORF among these still shorter than some ORFs we observe in our real DNA? If the real DNA ORFs are significantly longer than what we see in the garbage sequence, that is a very strong indicator that we did in fact find genes in our original DNA! Our first task is to write a function longestORFNoncoding (DNA, numReps) that makes a bunch of garbage sequences, finds the very longest ORF in all of these, and returns its length. Note: this function returns a number rather than a DNA string. OK, so now it's time to generate garbage sequences. We could generate totally random strings of the same length as our DNA string, but that might not be a very accurate test since our DNA string might have more nucleotides of one type and fewer of another. To be fair, our garbage strings should have the same nucleotides but just reordered or "shuffled" randomly. To do that, you'll first need to take your DNA string and turn it into a list of its constituent symbols. There is a built-in function called list that takes as input a string and returns the list of symbols in that string. >>> myList - list("hello") >>> myList ('h', 'e', 'l', 'l', 'o'l Now, in the random package (get it by including import random at the top of your file) there is a function called shuffle that scrambles the list. Unlike other functions that we've used until now, this shuffle function doesn't return a new list, but rather it actually changes the list that you gave it. Here's an example: >>> import random >>> myList = list("hello") >>> myList I'h', 'e', 'l', 'l', 'o'] >>> random.shuffle (myList) >>> myList >> collapse(['o', 'l', 'h', 'e', '1'1). 'olhel' For each garbage sequence you make, calculate the longest ORF using longestORFBothStrands. You should repeat this process numReps times, and return a number indicating the length of the longest ORF you see in al those repetitions. You can test your longestORFNoncoding function on our real Salmonella sequence >>> X73525-loadSeq("X73525.fa") Now run longestORFNoncoding a few times. Note that because it makes use of randomness, it will not give you exactly the same number each time. It will, however, be consistent enough for our purposes. >>> longestORFNoncoding(X73525,50) 624 >>> longestORFNoncoding(X73525,50) 693 Part 4: findORFs Our next step is to write a function findORFs (DNA) that will identify all the ORFs in the real (unshuffled) DNA and return them as a list. If there are none, it should return an empty list. Once again, this task is easy because we can make use of functions we have already written, findORFs should call oneFrameV2 in each of the three possible reading frames of the sequence. It should then combine all of the ORFs found in each frame and return them. Note that this strategy makes it possible to find overlapping reading frames. findORFs will likely be very similar to the longestORF that you wrote above. >>> findORFs ("ATGGGATGAATTAACCATGCCCTAA") CFB Web > Parti > Gene Finder De very similar to the longestORF that you wrote above. >>> findORFs ("ATGGGATGAATTAACCATGCCCTAA") ['ATGGGA', 'ATGCCC', 'ATGAAT'] >>> findORFs ("GGAGTAAGGGGG") (1 Part 5: findORFsBothStrands Next write a function called findORFsBothStrands DNA ) that searches both the forward and reverse complement strands for ORFs and returns a list with all the ORFs found. For example: >>> findORFsBothstrands ('ATGAAACAT') ("ATGAAACAT', 'ATGTTTCAT') Part 6: get Coordinates The functions we've written so far return the sequence of an ORF. However another bit of information we might like to know is its coordinates in the original DNA sequence. We'll now write a function getCoordinates (orf, DNA) which returns the beginning and end coordinates of an ORF in DNA. We will follow the convention of reporting everything in forward strand coordinates (even if the ORF is actually on the reverse complemented strand). First consider the case where the ORF falls on the forward (non reverse complemented) strand. In that case, we can obtain its beginning coordinate with the following syntax: >>> testDNA="ACGTTCGA" >>> testORF-"GTT" >>> testDNA.find(testORF) We can then get the end coordinate by adding the length of the ORF to the start coordinate. But what if the ORF is actually on the reverse complemented strand? Notice what happens when we use .find with a sequence that is not present: >>> testDNA-"ACGTTCGA" >>> testORP="GAA" >>> testDNA.find(testORF) -1 The method returns a -1. If we search the forward strand of DNA with an ORF and get a -1, then we should try searching with the reverse complement of orf. In our example, that would look like this: >>> testDNA="ACGTTCGA" >>> revCompTestORF=reverseComplement("GAA") >>> testDNA.find(revCompTestORF) Here are some examples of get Coordinates: >>> get Coordinates ("GTT", "ACGTTCGA) 112, 5 CFB Web > Parti > GeneFinder testuk="GAA >>> testDNA.find(testORF) -1 The method returns a -1. If we search the forward strand of DNA with an ORF and get a -1, then we should try searching with the reverse complement of orf. In our example, that would look like this: >>> testDNA-"ACGTTCGA" >>> revCompTestORF=reverseComplement("GAA") >>> testDNA.find(revCompTestORF) Here are some examples of get Coordinates: >>> get Coordinates ("TT", "ACGTTCGA) [2, 5) >>> getCoordinates (CGAA", "ACGTTCGA) [3, 7] Part 7: Gene finding at last! Write a function called geneFinder (DNA, minLen) that identifies ORFs longer than minLen, and returns a list with information about each. geneFinder should first call findORFsBothStrands to obtain a list of ORFs in the input DNA. It should then run through this list, keeping only those ORFs which are longer than minLen. For each ORF which is long enough, geneFinder should calculate 1. The beginning and end positions of the ORF in DNA using getCoordinates 2. The protein sequence of the ORF using codingStrandTOAA These should then be placed in a list: (beginningCoord, endCoord, proteinsequence) There will be a list like this for every ORF that is long enough. You will collect these lists in another list (a list of lists). A final step is to sort this list of lists before returning it. Say our final list of lists is called finaloutputList. Its elements are lists of this type: [beginningCoord, endCoord, proteinSequence). We can sort finaloutputList by beginning coordinate, and return it like this: finaloutputList.sort() return finaloutputList Part 8: Using our gene finder Now its time to apply our method to data and see what we get. 1. A. Load x73525. fa into python >>> X73525 = loadSeg("x73525.fa") and apply our gene-finding strategy to it. The first step is to decide on a threshold for geneFinder. Our strategy CFB Web > Parti > GeneFinder Part 8: Using our gene finder Now its time to apply our method to data and see what we get. 1. A. Load x73525. fa into python >>> X73525 = loadSeq("x73525.fa") and apply our gene-finding strategy to it. The first step is to decide on a threshold for geneFinder. Our strategy is to determine what the longest ORF we see in noncoding sequence is, and then to define ORFs longer than this as putative genes. Run longestORFNoncoding(X73525, 1500). This should be a relatively conservative way to pick a threshold. Now run geneFinder using the threshold value you get for minLen. >>> geneList = geneFinder (X73525, put your minLen_value_here Next write a short function printGenes (genelist) which prints the output of geneFinder in a nice human-readable form. Use it to print your results. Note that our gene-finding strategy is very simple, and is also relatively conservative. As a result, we are likely to miss some true genes which are short. But we hope that the genes our method does find are real ones. Paste your print Genes output into a file called GeneFinder.txt. B. Pick one of the genes from your output and blast its protein sequence. To start, open this link in a separate window: NCBI Blast. (On a mac, you can do that by holding down the "control" key while clicking on the link. You'll then have the option to open the link in a new window.) Go down to Basic Blast and follow the protein blast link. There should be a large box into which you can now input a protein ence. Then scroll down to the bottom of the page and click the "Blast" button. When you blast a sequence, the application carries out a search through its databases for known sequences that either match your sequence or are close to it, and returns those to you in a list. The BLAST output page will contain some graphics at the top. Scroll down past these to the section labelled "Descriptions". The first links in this section will be the closest sequences BLAST could find to your original input. Clicking on one of these will take you to a page with information about that sequence, including the name and what organism it can be found in. The right pane on that page has many useful links. Based on what you learn from these top BLAST hits, briefly describe the likely function of your gene at the top of GeneFinder.txt. 2. The famous biologist, Professor P.I. Pette, has been studying Next write a short function printGenes (geneList) which prints the output of genelinder in a nice human-readable form. Use it to print your results. Note that our gene-finding strategy is very simple, and is also relatively conservative. As a result, we are likely to miss some true genes which are short. But we hope that the genes our method does find are real ones. Paste your print Genes output into a file called GeneFinder.txt. B. Pick one of the genes from your output and blast its protein ence. To start, open this link in a separate window: NCBI Blast. (On a mac, you can do that by holding down the "control" key while clicking on the link. You'll then have the option to open the link in a new window.) Go down to Basic Blast and follow the protein blast link. There should be a large box into which you can now input a protein sequence. Then scroll down to the bottom of the page and click the "Blast" button. When you blast a sequence, the application carries out a search hrough its databases for known sequences that either match your sequence or are close to it, and returns those to you in a list. The BLAST output page will contain some graphics at the top. Scroll down past these to the section labelled "Descriptions". The first links in this section will be the closest sequences BLAST could find to your original input. Clicking on one of these will take you to a page with information about that sequence, including the name and what organism it can be found in. The right pane on that page has many useful links. Based on what you learn from these top BLAST hits, briefly describe the likely function of your gene at the top of GeneFinder.txt. The famous biologist, Professor P.I. Pette, has been studying Salmonella in her lab. She has obtained the following sequence from a novel pathogenic strain: salDNA.fa. Prof. Pette obtained this from a region unique to Salmonella. She believes it is involved in pathogenisis, and may be from pathogenicity island SPI1. Download this file and place it in the same directory as GeneFinder.py. Then use your code to help Prof Pette determine the function of this sequence. Identify any putative genes, and then blast them at NCBI BLAST. Note you will need to run longestORF Noncoding on this sequence to determine a good threshold for it. Using the links which NCBI BLAST gives you determine the name and function of your mystery gene(s). In GeneFinder.txt write a paragraph explaining what you have found. Gene finder All of your code for this problem should be in a file called GeneFinder.py. At the end you will also make a file with comments called GeneFinder.txt. Introduction In the text we discussed some investigations of Salmonella pathogenisis. Researchers identified genomic regions present in Salmonella but not in closely related species such as E. coli. Further study revealed that some of these regions are essential to Salmonella-specific pathogenic processes. One particular region was found to be needed for the entry of the bacterium into the epithelial cells of the gut. The researchers studying it wished to identify the protein-coding genes in it, so they sequenced it and analyzed that sequence for genes. In this homework, you will develop a simple gene finder, and use it to examine this sequence. You will also examine a related "mystery" sequence, and try to determine its function. You can begin by creating a homework file called GeneFinder.py. You will make use of a number of functions we have already written. You can give yourself access to the functions in your load.py and dna.py files by making sure they are present in the same directory as GeneFinder.py. Then include this at the top of GeneFinder.py: from load import * from dna import * Next, you should download the salmonella sequence we discussed in the text, accession number X73525, from NCBI as a fasta file. Go here for a reminder of how to do this. The file should be called x73525. fa (rename it if it's not). Move it to the same directory as your geneFinder.py file. A simple gene-finding strategy A simple gene-finding strategy is to look for large open reading frames. As we've seen, an open reading frame (ORF) is the stretch of sequence between a start codon and the next in frame stop codon. This strategy depends on the assumption that protein-coding sequence has larger ORFS than noncoding sequence. In noncoding regions "Start" and "Stop" codons appear by random chance, and the ORFs between them are not long. In many circumstances protein-coding genes have ORFs that are longer than this. To look for genes in a particular sequence, you can identify its longest ORFs. But how do you know if these are genes? To help decide, you can look at many known noncoding regions to obtain a distribution of open reading frame lengths for noncoding DNA. You can then compare the longest ORFs in our test sequence with this distribution. If the test sequence ORFs are longer, they are likely to be genes. In this problem the approach you will take is the following. You will compare open reading frames in our Salmonella sequence with open sequence ORFs are longer, they are likely to be genes. In this problem the approach you will take is the following. You will compare open reading frames in our Salmonella sequence with open reading frame lengths from known noncoding sequences. How do you get known noncoding sequences? One way is to make them randomly. Here you will do this by randomly shuffling (reordering) our genomic sequences More on that shortly! Part 1: Retrofitting functions from orf.py For your orf.py homework you created three functions. The first of these, restOfORP (DNA), can be used as written. Copy and paste this into your present homework file. Next you will write a modified version of oneFrame called oneFrameV2 (DNA). In our previous version of oneFrame, we searched the given reading frame for every start codon, and returned the ORF corresponding to it. This sometimes results in nested ORFs, and can be seen in the following example: >>> oneFrame("ATGCCCATGGGGAAATTTTGACCC") ['ATGCCCATGGGGAAATTT', 'ATGGGGAAATTT') Both ORFs end in the same stop codon. The second ORF is shorter, because it begins with an ATG which is internal to the first ORF. For our gene-finder application it is desirable to instead only return the largest ORF from a set of nested ORFs like this. >>> oneFrameV2 ("ATGCCCATGGGGAAATTTTGACCC") ('ATGCCCATGGGGAAATTT' ) A natural approach to writing oneFrameV2 is to search through the string till you find the first ATG. Then call restOFORF to get the ORF. But when you resume searching for the next ATG, you skip ahead and start looking after the end of the ORF you just found. A while loop will be very convenient here! Finally, create a longestORFV2 (DNA) function which calls oneFramev2. Part 2: longestORFBoth Strands(DNA) We are given a DNA sequence in 5' to 3' order. A gene might appear on this strand or its reverse complement. Thus, our next function is a very short one called longestORFBothStrands (DNA). This function takes a DNA string as input and finds the longest ORF on that DNA string or its reverse complement. This won't be hard! You can use the longestORFV2 function you have already written. First ask it for the longest ORF in the given DNA and then ask it for the longest ORF on its reverse complement (use your reverseComplement function to find the reverse complement). The longer of those two is the longest ORF possible (break ties arbitrarily). For example, >>> longestORFBothStrands('CTATTTCATG') ATGAAA Pause here to make sure you understand how we got that answer. Part 3: longestORFNoncoding (DNA, numReps) To assess whether long ORFs are genes, a researcher would ask the question, "is this sequence length indicative of a coding region or would I expect to see sequences this long in garbage?" By "garbage", of course, we mean just random sequences of nucleotides. We'll test this by generating a bunch of "garbage" sequences of the same length as our test DNA sequence, and measuring the maximum ORF length in each. Then, we'll ask the following question. Is the very longest ORF among these still shorter than some ORFs we observe in our real DNA? If the real DNA ORFs are significantly longer than what we see in the garbage sequence, that is a very strong indicator that we did in fact find genes in our original DNA! Our first task is to write a function longestORFNoncoding (DNA, numReps) that makes a bunch of garbage sequences, finds the very longest ORF in all of these, and returns its length. Note: this function returns a number rather than a DNA string. OK, so now it's time to generate garbage sequences. We could generate totally random strings of the same length as our DNA string, but that might not be a very accurate test since our DNA string might have more nucleotides of one type and fewer of another. To be fair, our garbage strings should have the same nucleotides but just reordered or "shuffled" randomly. To do that, you'll first need to take your DNA string and turn it into a list of its constituent symbols. There is a built-in function called list that takes as input a string and returns the list of symbols in that string. >>> myList - list("hello") >>> myList ('h', 'e', 'l', 'l', 'o'l Now, in the random package (get it by including import random at the top of your file) there is a function called shuffle that scrambles the list. Unlike other functions that we've used until now, this shuffle function doesn't return a new list, but rather it actually changes the list that you gave it. Here's an example: >>> import random >>> myList = list("hello") >>> myList I'h', 'e', 'l', 'l', 'o'] >>> random.shuffle (myList) >>> myList >> collapse(['o', 'l', 'h', 'e', '1'1). 'olhel' For each garbage sequence you make, calculate the longest ORF using longestORFBothStrands. You should repeat this process numReps times, and return a number indicating the length of the longest ORF you see in al those repetitions. You can test your longestORFNoncoding function on our real Salmonella sequence >>> X73525-loadSeq("X73525.fa") Now run longestORFNoncoding a few times. Note that because it makes use of randomness, it will not give you exactly the same number each time. It will, however, be consistent enough for our purposes. >>> longestORFNoncoding(X73525,50) 624 >>> longestORFNoncoding(X73525,50) 693 Part 4: findORFs Our next step is to write a function findORFs (DNA) that will identify all the ORFs in the real (unshuffled) DNA and return them as a list. If there are none, it should return an empty list. Once again, this task is easy because we can make use of functions we have already written, findORFs should call oneFrameV2 in each of the three possible reading frames of the sequence. It should then combine all of the ORFs found in each frame and return them. Note that this strategy makes it possible to find overlapping reading frames. findORFs will likely be very similar to the longestORF that you wrote above. >>> findORFs ("ATGGGATGAATTAACCATGCCCTAA") CFB Web > Parti > Gene Finder De very similar to the longestORF that you wrote above. >>> findORFs ("ATGGGATGAATTAACCATGCCCTAA") ['ATGGGA', 'ATGCCC', 'ATGAAT'] >>> findORFs ("GGAGTAAGGGGG") (1 Part 5: findORFsBothStrands Next write a function called findORFsBothStrands DNA ) that searches both the forward and reverse complement strands for ORFs and returns a list with all the ORFs found. For example: >>> findORFsBothstrands ('ATGAAACAT') ("ATGAAACAT', 'ATGTTTCAT') Part 6: get Coordinates The functions we've written so far return the sequence of an ORF. However another bit of information we might like to know is its coordinates in the original DNA sequence. We'll now write a function getCoordinates (orf, DNA) which returns the beginning and end coordinates of an ORF in DNA. We will follow the convention of reporting everything in forward strand coordinates (even if the ORF is actually on the reverse complemented strand). First consider the case where the ORF falls on the forward (non reverse complemented) strand. In that case, we can obtain its beginning coordinate with the following syntax: >>> testDNA="ACGTTCGA" >>> testORF-"GTT" >>> testDNA.find(testORF) We can then get the end coordinate by adding the length of the ORF to the start coordinate. But what if the ORF is actually on the reverse complemented strand? Notice what happens when we use .find with a sequence that is not present: >>> testDNA-"ACGTTCGA" >>> testORP="GAA" >>> testDNA.find(testORF) -1 The method returns a -1. If we search the forward strand of DNA with an ORF and get a -1, then we should try searching with the reverse complement of orf. In our example, that would look like this: >>> testDNA="ACGTTCGA" >>> revCompTestORF=reverseComplement("GAA") >>> testDNA.find(revCompTestORF) Here are some examples of get Coordinates: >>> get Coordinates ("GTT", "ACGTTCGA) 112, 5 CFB Web > Parti > GeneFinder testuk="GAA >>> testDNA.find(testORF) -1 The method returns a -1. If we search the forward strand of DNA with an ORF and get a -1, then we should try searching with the reverse complement of orf. In our example, that would look like this: >>> testDNA-"ACGTTCGA" >>> revCompTestORF=reverseComplement("GAA") >>> testDNA.find(revCompTestORF) Here are some examples of get Coordinates: >>> get Coordinates ("TT", "ACGTTCGA) [2, 5) >>> getCoordinates (CGAA", "ACGTTCGA) [3, 7] Part 7: Gene finding at last! Write a function called geneFinder (DNA, minLen) that identifies ORFs longer than minLen, and returns a list with information about each. geneFinder should first call findORFsBothStrands to obtain a list of ORFs in the input DNA. It should then run through this list, keeping only those ORFs which are longer than minLen. For each ORF which is long enough, geneFinder should calculate 1. The beginning and end positions of the ORF in DNA using getCoordinates 2. The protein sequence of the ORF using codingStrandTOAA These should then be placed in a list: (beginningCoord, endCoord, proteinsequence) There will be a list like this for every ORF that is long enough. You will collect these lists in another list (a list of lists). A final step is to sort this list of lists before returning it. Say our final list of lists is called finaloutputList. Its elements are lists of this type: [beginningCoord, endCoord, proteinSequence). We can sort finaloutputList by beginning coordinate, and return it like this: finaloutputList.sort() return finaloutputList Part 8: Using our gene finder Now its time to apply our method to data and see what we get. 1. A. Load x73525. fa into python >>> X73525 = loadSeg("x73525.fa") and apply our gene-finding strategy to it. The first step is to decide on a threshold for geneFinder. Our strategy CFB Web > Parti > GeneFinder Part 8: Using our gene finder Now its time to apply our method to data and see what we get. 1. A. Load x73525. fa into python >>> X73525 = loadSeq("x73525.fa") and apply our gene-finding strategy to it. The first step is to decide on a threshold for geneFinder. Our strategy is to determine what the longest ORF we see in noncoding sequence is, and then to define ORFs longer than this as putative genes. Run longestORFNoncoding(X73525, 1500). This should be a relatively conservative way to pick a threshold. Now run geneFinder using the threshold value you get for minLen. >>> geneList = geneFinder (X73525, put your minLen_value_here Next write a short function printGenes (genelist) which prints the output of geneFinder in a nice human-readable form. Use it to print your results. Note that our gene-finding strategy is very simple, and is also relatively conservative. As a result, we are likely to miss some true genes which are short. But we hope that the genes our method does find are real ones. Paste your print Genes output into a file called GeneFinder.txt. B. Pick one of the genes from your output and blast its protein sequence. To start, open this link in a separate window: NCBI Blast. (On a mac, you can do that by holding down the "control" key while clicking on the link. You'll then have the option to open the link in a new window.) Go down to Basic Blast and follow the protein blast link. There should be a large box into which you can now input a protein ence. Then scroll down to the bottom of the page and click the "Blast" button. When you blast a sequence, the application carries out a search through its databases for known sequences that either match your sequence or are close to it, and returns those to you in a list. The BLAST output page will contain some graphics at the top. Scroll down past these to the section labelled "Descriptions". The first links in this section will be the closest sequences BLAST could find to your original input. Clicking on one of these will take you to a page with information about that sequence, including the name and what organism it can be found in. The right pane on that page has many useful links. Based on what you learn from these top BLAST hits, briefly describe the likely function of your gene at the top of GeneFinder.txt. 2. The famous biologist, Professor P.I. Pette, has been studying Next write a short function printGenes (geneList) which prints the output of genelinder in a nice human-readable form. Use it to print your results. Note that our gene-finding strategy is very simple, and is also relatively conservative. As a result, we are likely to miss some true genes which are short. But we hope that the genes our method does find are real ones. Paste your print Genes output into a file called GeneFinder.txt. B. Pick one of the genes from your output and blast its protein ence. To start, open this link in a separate window: NCBI Blast. (On a mac, you can do that by holding down the "control" key while clicking on the link. You'll then have the option to open the link in a new window.) Go down to Basic Blast and follow the protein blast link. There should be a large box into which you can now input a protein sequence. Then scroll down to the bottom of the page and click the "Blast" button. When you blast a sequence, the application carries out a search hrough its databases for known sequences that either match your sequence or are close to it, and returns those to you in a list. The BLAST output page will contain some graphics at the top. Scroll down past these to the section labelled "Descriptions". The first links in this section will be the closest sequences BLAST could find to your original input. Clicking on one of these will take you to a page with information about that sequence, including the name and what organism it can be found in. The right pane on that page has many useful links. Based on what you learn from these top BLAST hits, briefly describe the likely function of your gene at the top of GeneFinder.txt. The famous biologist, Professor P.I. Pette, has been studying Salmonella in her lab. She has obtained the following sequence from a novel pathogenic strain: salDNA.fa. Prof. Pette obtained this from a region unique to Salmonella. She believes it is involved in pathogenisis, and may be from pathogenicity island SPI1. Download this file and place it in the same directory as GeneFinder.py. Then use your code to help Prof Pette determine the function of this sequence. Identify any putative genes, and then blast them at NCBI BLAST. Note you will need to run longestORF Noncoding on this sequence to determine a good threshold for it. Using the links which NCBI BLAST gives you determine the name and function of your mystery gene(s). In GeneFinder.txt write a paragraph explaining what you have found
Step by Step Solution
There are 3 Steps involved in it

Get step-by-step solutions from verified subject matter experts