

Answered step by step
Verified Expert Solution
Question
1 Approved Answer
Python computer program. CSE 231 Spring 2018 Programming Project 06 Edit 2/21: removed one line from Function Test 2 to improve clarity This assignment is
Python computer program. 




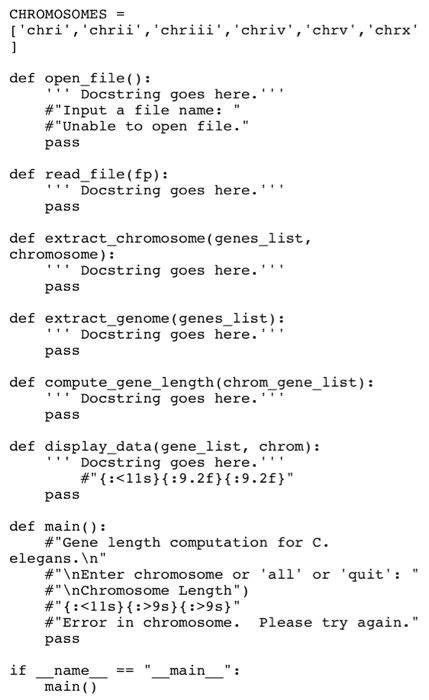
CSE 231 Spring 2018 Programming Project 06 Edit 2/21: removed one line from Function Test 2 to improve clarity This assignment is worth 45 points and must be completed and turned in before 11:59 on Monday February 26, 2018. Assignment Overview This assignment will give you more experience on the use of 1. Lists and tuples 2. function 3. File manipulation The goal of this project is to extract gene lengths from a gene annotation file. With a gene annotation GFF file, you will nced to extract the gene coordinates on each chromosome and calculate the average and standard deviation of gene lengths. Assignment Background The cukaryotic genome is composed of multiple chromosomes. On cach chromosome, there are multiple genes. In bioinformatics, the genome annotations can be saved in a file format called GFF. In NCBI genome database (https://www.ncbi.nlm.nih.gov/genome), there are many publically available annotated organisms. These annotated genomes can be downloaded in multiple file formats, including GFF format. For this project, we will focus on a relatively simple model species: Cacnorhabditis elegans. This worm has a genome of six chromosomes named chrl, chrll, chrill, chrlV,chrV, and chrX. We provide two input files: c.elegans_small.gff C.elegans.gff a small file for development a real BIG data file Project Deseription a) open file) prompts the user to enter a filename. The program will try to open a tab- separated GFF file (a text file). An error message should be shown if the file cannot be opened. This function will loop until it receives proper input and successfully opens the file. It retums a file pointer. b) read file(fp) receivers a file pointer of the data file and read all the genes information. For this project, we are only interested in the following columns: the chromosome name (string) is in column 0, the gene start is in column 3, and the gene end is in column 4. Convert number strings to int. No other values are needed for this project. If a value is missing, use 0 as the value. For each gene, save it in a tuple, (chromosome. gene start. gene end). and append each tuple to a list of genes. Sort the list and then return the sorted list of genes (sorting makes a canonical list for comparison testing on Mimir). b) extract_chromosome (genes list, chromosome) receives a list of genes (such as what was retuned by the read file) function) and a chromosome name, extract the gene information for this chromosome and save in list chrom_gene list. Sort and return the list (sorting makes a canonical list for comparison testing on Mimir) c) extract genome (genes_list) receives a list of genes and extract the gene information for each chromosome. In this function, use extract_chromosome(genes list, chromosome) to extract Step by Step Solution
There are 3 Steps involved in it
Step: 1

Get Instant Access to Expert-Tailored Solutions
See step-by-step solutions with expert insights and AI powered tools for academic success
Step: 2

Step: 3
Ace Your Homework with AI
Get the answers you need in no time with our AI-driven, step-by-step assistance
Get Started
Information Modeling And Relational Databases
Authors: Terry Halpin, Tony Morgan
2nd Edition
0123735688, 978-0123735683