Python - This is my code so far ^
I need help with part 2:



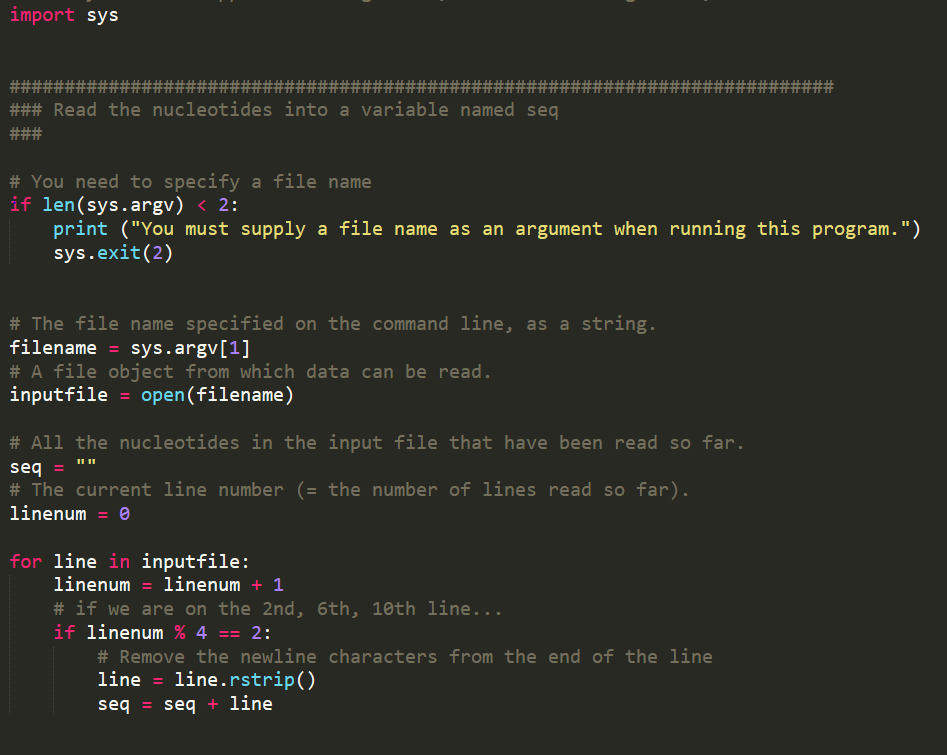
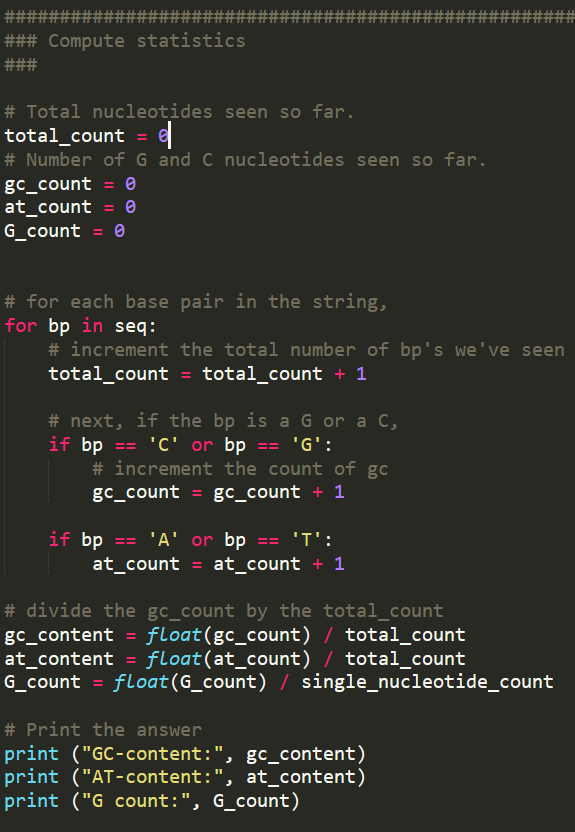
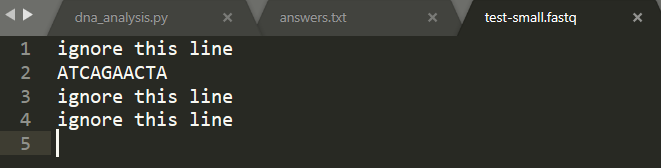
import sys ### Read the nucleotides into a variable named sea # You need to specify a file name if len(sys.argv) 2: print ("You must supply a file name as an argument when running this program.") sys.exit(2) # The file name specified on the command line, as a string filename sys.argv[1] # A file object from which data can be read . inputfile -open (filename) # All the nucleotides in the input file that have been read so far. seq"" # The current line number (-the number of lines read so far). linenum for line in inputfile: linenumlinenum 1 # if we are on the 2nd, 6th, 10th line.. if linenum % 4 =: 2: # Remove the newline characters from the end of the line line line.rstrip) seq seq line ### Compute statistics # Total nucleotides seen so far. total count - # Number of G and C nucleotides seen so far. gc_count at count G count # for each base pair in the string, for bp in seq: # increment the total number of bp's we've seen total count total count 1 # next, if the bp is a G or a C, if bp'C' or bp'G' # increment the count of gc gc_count gc_count+ 1 at countat count1 # divide the gc count by the total count gc_content float (gc_count) /total_count at_content float (at_count) /total_count G_count-float (G_count) / single_nucleotide_count # Print the answer print ("GC-content:", gc_content) print ("AT-content:", at_content) print ("G count:", G_count) dna_analysis.py answers.txt test-small.fastq 1 ignore this line 2 ATCAGAACTA 3 ignore this line 4 ignore this line 5 Part 2a: Count nucleotides Augment your program so that it also computes and prints the number of A nucleotides, the number of T nucleotides, the number of G nucleotides, and the number of C nucleotides. When doing this, add at most one extra loop to your program. You can solve this part without adding any new loops at all, by reusing an existing loop Check your work by manually computing the results for file test-small.fastq, then comparing them to the output of running your program on test-small.fastq Run your program on sample_1.fastq. Cut-and-paste the relevant lines of output into answers.txt (the lines that indicate the G count, C count, A count, and T count) Part 2b: Sanity-check the data For each of the 11 .fastq files, compare the following three quantities: the sum of the A count, the C count, the G count, and the T count the total_count variable the length of the seq variable. You can compute this with len(seq) In other words, compute the three numbers for test-small.fastq and determine whether they are equal or different. Then do the same for test-high-gc-1.fastq etc For at least one file, at least two of these metrics will differ. In your answers.txt file, state which file(s) and which metrics. (If all the metries are equal for each file, then your code contains a mistake.) In your answers.txt file, write a short paragraph that explains why Explaining why (or debugging your code if all the metrics were the same) might require you to do some detective work. For instance, to understand the issue, you may need to load a file into a text editor and examine it. We strongly suggest that you start with the smallest file for which the numbers are not all the same. Perusal of the file may help you. Failing that, you can manually compute each of the counts, and then compare your manual results to what your program computes to determine where the error lies. A final approach would be to modify your program, or create a new program, to compute the three metrics for each line of a file separately: if the metrics differ for an entire file, then they must differ for some specific line, and then examining that line will help you understand the problem. If all of the three quantities that you measured in Part2b are the same, then it would not matter which one you used in the denominator when computing the GC content. In fact, you saw that the numbers are not the same. In file answers.txt, state which of these quantities can be used in the denominator and which cannot, and why If your program incorrectly computed the GC content (which should be equal to (G+C)/CA+C+G+T)), then state that fact in your answers.txt file. Then, go back and correct it, and also update any incorrect answers elsewhere in your answers.txt file Part 2c: Compute the AT/GC ratio Sometimes biologists use the AT/GC ratio, defined as (A+T)/(G+C), rather than the GC-content, which is defined as (G+C)/(A+C+G+T). Modify your program so that it also computes the AT/GC ratio Check your work by manually computing the results for file test-small.fastq. Compare them to the output of running your program on test-small.fastq Run your program on sample_1.fastq. Cut-and-paste the relevant lines of output into answers.txt (the line that indicates the AT/GC ratio) Part 2d: Categorize organisms The GC content can be used to categorize microorganisms. Modify your program to print out a classification of the organism in the file. If the GC content is above 60%, the organism is considered "high GC content". If the GC content is below 40%, the organism is considered "low GC content". Otherwise, the organism is considered "moderate GC content" Biologists can use GC content for classifying species, for determining the melting temperature of the DNA (useful for both ecology and experimentation, for example PCR is more difficult on organisms with high GC content), and for other purposes. Here are some examples: The GC content of Streptomyces coelicolor A3(2) is 72% The GC content of Yeast (Saccharomyces cerevisiae) is 38%. The GC content of Thale Cress (Arabidopsis thaliana) is 36%. The GC content of Plasmodium falciparum is 20%. Again, test your work. The test-small.fastq file has low GC content. We have provided four other test files, whose names explain their GC content: test-moderate- gc-1.fastq, test-moderate-gc-2.fastq, test-high-gc-1.fastq, test-high-gc-2.fastq After your program works for all the test files, run it on sample_1.fastq. Cut-and-paste just the relevant line of output from your program into answers.txt




