

Question: the required functions are at the end for easy use. These are the functions required that I have coded. I am lost on how to
the required functions are at the end for easy use. 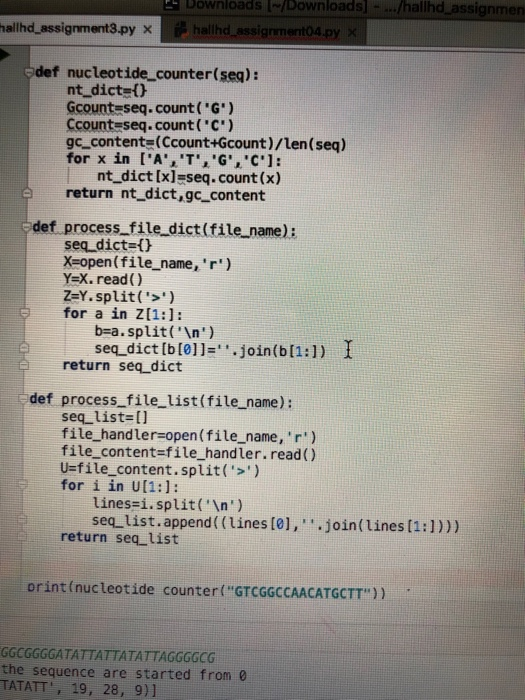
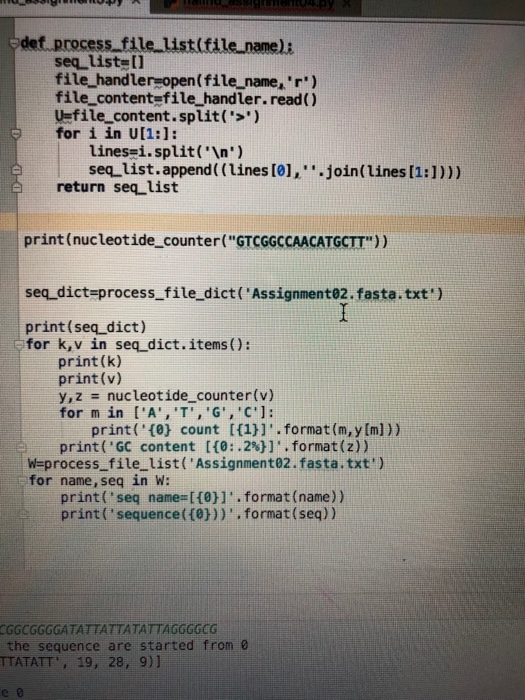 These are the functions required that I have coded. I am lost on how to contribute them into these new functions.
These are the functions required that I have coded. I am lost on how to contribute them into these new functions.


Once the new functions are coded they need to be printed out like the pictures below. I keep recieving many errors with my codes while trying these.

here are the codes I coded. I don't know how else to share them.
def nucleotide_counter(seq): nt_dict={} Gcount=seq.count('G') Ccount=seq.count('C') gc_content=(Ccount+Gcount)/len(seq) for x in ['A','T','G','C']: nt_dict[x]=seq.count(x) return nt_dict,gc_content def process_file_dict(file_name): seq_dict={} X=open(file_name,'r') Y=X.read() Z=Y.split('>') for a in Z[1:]: b=a.split(' ') seq_dict[b[0]]=''.join(b[1:]) return seq_dict def process_file_list(file_name): seq_list=[] file_handler=open(file_name,'r') file_content=file_handler.read() U=file_content.split('>') for i in U[1:]: lines=i.split(' ') seq_list.append((lines[0],''.join(lines[1:]))) return seq_list print(nucleotide_counter("GTCGGCCAACATGCTT")) seq_dict=process_file_dict('Assignment02.fasta.txt') print(seq_dict) for k,v in seq_dict.items(): print(k) print(v) y,z = nucleotide_counter(v) for m in ['A','T','G','C']: print('{0} count [{1}]'.format(m,y[m])) print('GC content [{0:.2%}]'.format(z)) W=process_file_list('Assignment02.fasta.txt') for name,seq in W: print('seq name=[{0}]'.format(name)) print('sequence({0}))'.format(seq)) codon sequence = ATGC
Downloads i~ / Downloads ] -/hallhd_ass allhd_assignment3py hallhd assignment04.p def nucleotide counter(seq): nt dict ) Gcount seq.count ('G) ccount-seq. count ( " " ) gc contents(Ccount+Gcount)/len(seq) for x in ['A T 'G, c1: nt-dict [x] =seq. count (x) ereturn nt_dict,gc content def process file dict(file name): seq dict X open(file_name,'r) Y-. read ( ) . split('>' ) for a in Z[1:1: b-a.split('n) seq, dict [b [0] ]-'''join ( b [1:1) i return seq dict def process file list(file name): seq-list=[] file handler open(file name, 'r) file_content-file handler. read) U-file_content.split(>') for i in U[1:]: lines-i.split('n') seq_ list.append( (lines tel,' ".join(lines [1:1))) return seq list orint(nucleotide counter("GTCGGCCAACATGCTT")) GGCGGGGATATTATTATATTAGGGGCG the sequence are started from 0 TATATT, 19, 28, 9)1 Why this is important? This is because you want your program generic enough to process any given FASTA file. For this assignment, you need to keep the two functions that you coded for assignment No.3: nucleotide_counter(seq) and process_file_dictfile_name), with a minor change. Make sure that in the main part of your program, the function call for process file_dict() must use a named string object as the argument, which will be provided by the above command line input (i.e., the FASTA file name). You cannot hard-code the file name (e.g., process file_ dict(filename) not process file_dict('Assignment04a.fasta'). Moreover, the dictionary returned by this function will have the sequence name as the key, a tuple of the description and sequence bases as the value (e.g., ('Huamn Breast Cancer Gene', 'GGAGGAGGA)) Here are the new functions that you need to code: # Given a DNA sequence in string object, this function will return its reverse # complementary sequence (a named string object). For example, ATGC will get GCAT ef rev_comp_seq(seq): rev_comp_seq return rev_comp_seq # Given a DNA sequence in string object, this function will return a dictionary that have all # def codon-usage(seq): codons existing in this sequence and the relevant frequency counts. You can utilize the last function that you coded for assignment No.2 and do some modifications. # codon_dict- return codon_dict def detect-homopolymer(seq): # Given a DNA sequence in string object, this function will detect all homopolymers with the minimum length of 8 or more (e.g. . CCCCCCCCCCC" or "AAAAAAAA') Consequently, we need to know the homopolymer sequence, start and end position. aswell as its length (e.g.,'ccccccccccc', start-5,end-15,length-10). All detected homopolymers will be saved into a list where each element of the list is a tuple that has 4 parts (e.g., ( 'ccccccccccc. , 5, 15, 10)) # # # # # homo_list-U return homo-list # The returned list is named as homo-list 14). # Given a DNA sequence in string object, this function will return a string of protein # sequence, using the following strategy: get the first 3 nucleotides as the first codon def translation(sea): " and then get the next 3 nucleotides as the second codon. Keep going until you get the # the last 3 nucleotides as the last codon. Make sure that you are clear any two # adjacent codons do not have any overlapped # dictionary that has codon (key) and amino acid (value) association to get protein # sequence. nucleotide. You need to design a genetic_code 0 return protein seq # a string object that represents the protein sequence for the given DNA sequence def six frame translation(seq): # Given a DNA sequence in string object, this function will not return anything, but it will # print out the 6 frame translation results (e.g., 6 proteins sequences). In this function # you can call translation() 6 times, obtain 6 protein sequences, and print them. Also, # you need to call rev-comp-seq() to get the reverse complementary sequence for the # given DNA sequence. def detect enzyme(seq, enzyme_dict) # Given a DNA sequence in string object and a dictionary that contains enzyme name # (key) and sequence (value), this function will not return anything, but it will # print out all the enzyme restriction sites, start and end positions, and lengths. # In the main part of your program, you need to generate the enzyme-dict dictionary # with the following keys and values. Then, you can use it as one of the function # arguments: GAATTC AAGCTT GCGGCC GGATCC ECORI Ht Hindil H Noti #BamHl # Inside this function, you need to declare a dictionary for the detected enzymes with enzyme name as the key and a list of the tuple (start, end, length) as the value. For one enzyme, you might find multiple occurrences, so each occurrence will be one detected-enzymes() # # # element in the list. Then, you navigate detect-enzyme to print out findings The 2nd sequence will need to be 2, the third one 3 (11 General Information Sequence Name: (-] Description: (1 Sequence: -1 Reverse Complenentary Sequence: () Sequence Length: [358] Nucleotide Counts (A-100, T-50, G-100, C108) Gc content-46.10 (2) 6-Frame Translation Frame (+1): MDCHPIWREVRYCEYPLHVEMACKR Prame (+2): MCHPTWREVRYCEYPLHVEMACKR Frame (+3): MHPINREVRYCEYPLHVEMACKR Frame (-1): MIWREVRYCEYPLHVEMACKRO Frame (-2): MRYCEYPLHVEMACKRLLLLLLLR Frame (-3): MDCHPINREVRYCEYPLHVEMAC (3) Codon Usage ATG 33 ACT 21 (4) Homopolymer Detection Homopol ymer End Length (5) Enzyme Detection Name EcoRI EcoRi NotI HindIII Sequence Start GAATTC GAATTC 381 Occurrence End 386 Except for functions detect enzymel) and six_frame translationl), all print statements for the required output should be within the main part of your program. While for computer science, index starts with 0, sequence position always start with 1 for biologists. What happens in the main part of your script? (1) Call the function process file_dict) by providing the required argument, which was provided by the command line input and obtain a named dictionary that contains both keys (sequence names) and values (descriptions and sequences as the tuple). 12) Now, you need to navigate each key of the dictionary and retrieve the corresponding values (ie, sequence (3) You might notice that all S functions, except the detect enzymel). take only one argument seq, that description and sequence base). Then, you can use call the functions that you code to get the proper print out sequence bases you obtained for each sequence. The function detect enzymel) requires two arguments enzyme_dict. Obviously, you need to create enzyme_ dict first and then call the function. seq and (4) Although we ask you not to have print statements within functions in order to get proper printout, except in detect_enzymel) and six_ frame_translationl), for debugging purpose you need to have proper print statements with any function. After debugging, make sure to comment these print statements out so that they will not affect your printout Downloads i~ / Downloads ] -/hallhd_ass allhd_assignment3py hallhd assignment04.p def nucleotide counter(seq): nt dict ) Gcount seq.count ('G) ccount-seq. count ( " " ) gc contents(Ccount+Gcount)/len(seq) for x in ['A T 'G, c1: nt-dict [x] =seq. count (x) ereturn nt_dict,gc content def process file dict(file name): seq dict X open(file_name,'r) Y-. read ( ) . split('>' ) for a in Z[1:1: b-a.split('n) seq, dict [b [0] ]-'''join ( b [1:1) i return seq dict def process file list(file name): seq-list=[] file handler open(file name, 'r) file_content-file handler. read) U-file_content.split(>') for i in U[1:]: lines-i.split('n') seq_ list.append( (lines tel,' ".join(lines [1:1))) return seq list orint(nucleotide counter("GTCGGCCAACATGCTT")) GGCGGGGATATTATTATATTAGGGGCG the sequence are started from 0 TATATT, 19, 28, 9)1 Why this is important? This is because you want your program generic enough to process any given FASTA file. For this assignment, you need to keep the two functions that you coded for assignment No.3: nucleotide_counter(seq) and process_file_dictfile_name), with a minor change. Make sure that in the main part of your program, the function call for process file_dict() must use a named string object as the argument, which will be provided by the above command line input (i.e., the FASTA file name). You cannot hard-code the file name (e.g., process file_ dict(filename) not process file_dict('Assignment04a.fasta'). Moreover, the dictionary returned by this function will have the sequence name as the key, a tuple of the description and sequence bases as the value (e.g., ('Huamn Breast Cancer Gene', 'GGAGGAGGA)) Here are the new functions that you need to code: # Given a DNA sequence in string object, this function will return its reverse # complementary sequence (a named string object). For example, ATGC will get GCAT ef rev_comp_seq(seq): rev_comp_seq return rev_comp_seq # Given a DNA sequence in string object, this function will return a dictionary that have all # def codon-usage(seq): codons existing in this sequence and the relevant frequency counts. You can utilize the last function that you coded for assignment No.2 and do some modifications. # codon_dict- return codon_dict def detect-homopolymer(seq): # Given a DNA sequence in string object, this function will detect all homopolymers with the minimum length of 8 or more (e.g. . CCCCCCCCCCC" or "AAAAAAAA') Consequently, we need to know the homopolymer sequence, start and end position. aswell as its length (e.g.,'ccccccccccc', start-5,end-15,length-10). All detected homopolymers will be saved into a list where each element of the list is a tuple that has 4 parts (e.g., ( 'ccccccccccc. , 5, 15, 10)) # # # # # homo_list-U return homo-list # The returned list is named as homo-list 14). # Given a DNA sequence in string object, this function will return a string of protein # sequence, using the following strategy: get the first 3 nucleotides as the first codon def translation(sea): " and then get the next 3 nucleotides as the second codon. Keep going until you get the # the last 3 nucleotides as the last codon. Make sure that you are clear any two # adjacent codons do not have any overlapped # dictionary that has codon (key) and amino acid (value) association to get protein # sequence. nucleotide. You need to design a genetic_code 0 return protein seq # a string object that represents the protein sequence for the given DNA sequence def six frame translation(seq): # Given a DNA sequence in string object, this function will not return anything, but it will # print out the 6 frame translation results (e.g., 6 proteins sequences). In this function # you can call translation() 6 times, obtain 6 protein sequences, and print them. Also, # you need to call rev-comp-seq() to get the reverse complementary sequence for the # given DNA sequence. def detect enzyme(seq, enzyme_dict) # Given a DNA sequence in string object and a dictionary that contains enzyme name # (key) and sequence (value), this function will not return anything, but it will # print out all the enzyme restriction sites, start and end positions, and lengths. # In the main part of your program, you need to generate the enzyme-dict dictionary # with the following keys and values. Then, you can use it as one of the function # arguments: GAATTC AAGCTT GCGGCC GGATCC ECORI Ht Hindil H Noti #BamHl # Inside this function, you need to declare a dictionary for the detected enzymes with enzyme name as the key and a list of the tuple (start, end, length) as the value. For one enzyme, you might find multiple occurrences, so each occurrence will be one detected-enzymes() # # # element in the list. Then, you navigate detect-enzyme to print out findings The 2nd sequence will need to be 2, the third one 3 (11 General Information Sequence Name: (-] Description: (1 Sequence: -1 Reverse Complenentary Sequence: () Sequence Length: [358] Nucleotide Counts (A-100, T-50, G-100, C108) Gc content-46.10 (2) 6-Frame Translation Frame (+1): MDCHPIWREVRYCEYPLHVEMACKR Prame (+2): MCHPTWREVRYCEYPLHVEMACKR Frame (+3): MHPINREVRYCEYPLHVEMACKR Frame (-1): MIWREVRYCEYPLHVEMACKRO Frame (-2): MRYCEYPLHVEMACKRLLLLLLLR Frame (-3): MDCHPINREVRYCEYPLHVEMAC (3) Codon Usage ATG 33 ACT 21 (4) Homopolymer Detection Homopol ymer End Length (5) Enzyme Detection Name EcoRI EcoRi NotI HindIII Sequence Start GAATTC GAATTC 381 Occurrence End 386 Except for functions detect enzymel) and six_frame translationl), all print statements for the required output should be within the main part of your program. While for computer science, index starts with 0, sequence position always start with 1 for biologists. What happens in the main part of your script? (1) Call the function process file_dict) by providing the required argument, which was provided by the command line input and obtain a named dictionary that contains both keys (sequence names) and values (descriptions and sequences as the tuple). 12) Now, you need to navigate each key of the dictionary and retrieve the corresponding values (ie, sequence (3) You might notice that all S functions, except the detect enzymel). take only one argument seq, that description and sequence base). Then, you can use call the functions that you code to get the proper print out sequence bases you obtained for each sequence. The function detect enzymel) requires two arguments enzyme_dict. Obviously, you need to create enzyme_ dict first and then call the function. seq and (4) Although we ask you not to have print statements within functions in order to get proper printout, except in detect_enzymel) and six_ frame_translationl), for debugging purpose you need to have proper print statements with any function. After debugging, make sure to comment these print statements out so that they will not affect your printout
Step by Step Solution
There are 3 Steps involved in it

Get step-by-step solutions from verified subject matter experts