

Answered step by step
Verified Expert Solution
Question
1 Approved Answer
The first two images are the templates, and 5 6 def read_one_seq_fasta (fasta_file): 7 8 9 10 11 12 13 14 15 16 def gc_content
The first two images are the templates, and 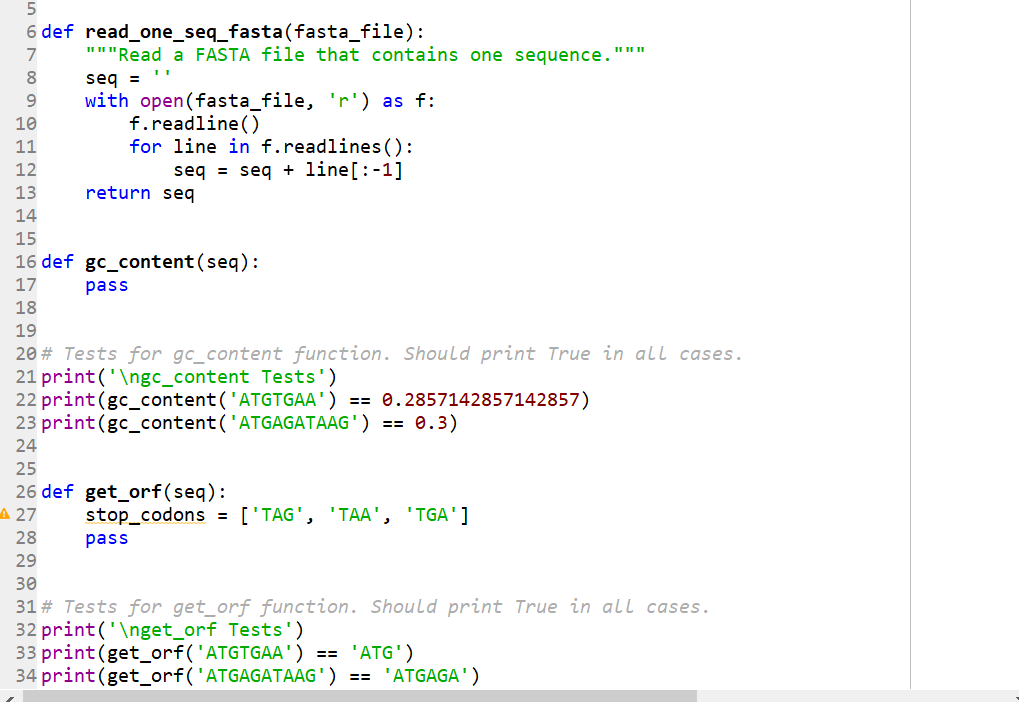
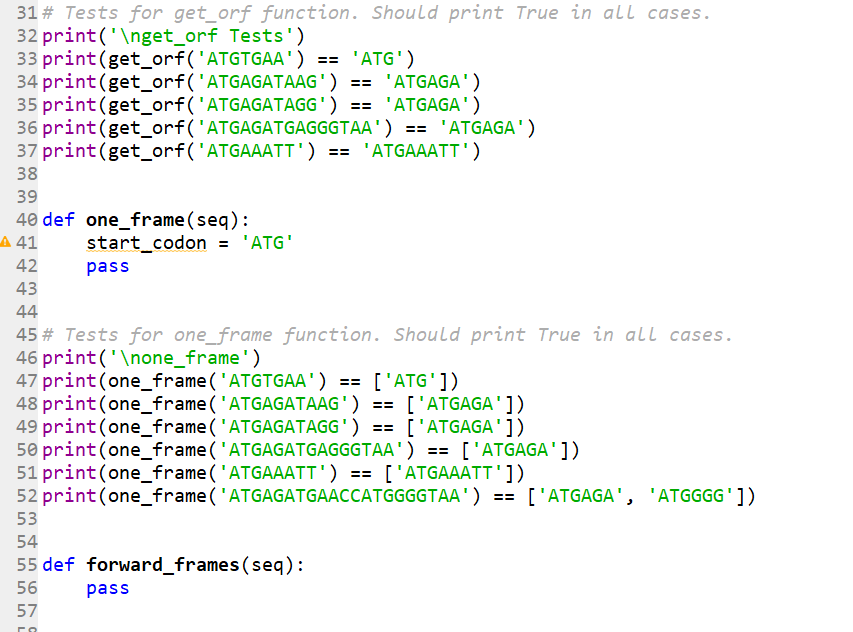


5 6 def read_one_seq_fasta (fasta_file): 7 8 9 10 11 12 13 14 15 16 def gc_content (seq): 17 pass """Read a FASTA file that contains one sequence. seq = with open(fasta_file, 'r') as f: f.readline() for line in f.readlines (): seq = seq + line[:-1] return seq 18 19 20 # Tests for gc_content function. Should print True in all cases. 21 print('gc_content Tests') 22 print (gc_content ('ATGTGAA') == 0.2857142857142857) 23 print (gc_content('ATGAGATAAG') == 0.3) 24 25 26 def get_orf(seq): A 27 || || || stop_codons = ['TAG', 'TAA', 'TGA'] pass 28 29 30 31# Tests for get_orf function. Should print True in all cases. 32 print('get_orf Tests') 33 print (get_orf('ATGTGAA') == 'ATG') 34 print (get_orf('ATGAGATAAG') == 'ATGAGA') 31# Tests for get_orf function. Should print True in all cases. 32 print('get_orf Tests') 33 print (get_orf('ATGTGAA') 34 print (get_orf('ATGAGATAAG') 35 print (get_orf('ATGAGATAGG') 36 print (get_orf('ATGAGATGAGGGTAA') == 'ATGAGA') 37 print (get_orf('ATGAAATT') == 'ATGAAATT') 38 39 40 def one_frame(seq): A 41 Inii in in in L 42 43 44 45# Tests for one_frame function. Should print True in all cases. 46 print('one_frame') 47 print (one_frame('ATGTGAA') == ['ATG']) 48 print (one_frame('ATGAGATAAG') == ['ATGAGA']) 49 print (one_frame('ATGAGATAGG') == ['ATGAGA']) 50 print (one_frame('ATGAGATGAGGGTAA') == ['ATGAGA']) 51 print (one_frame('ATGAAATT') == ['ATGAAATT']) 52 print 53 195 AWN 56 start_codon = 'ATG' 54 55 def forward_frames (seq): 57 ra pass == 'ATG') == 'ATGAGA') == 'ATGAGA') (one_frame('ATGAGATGAACCATGGGGTAA') == ['ATGAGA', 'ATGGGG']) pass To start, use the provided template file (on Blackboard): project_01_template.py. Replace the pass statements with your code. Notice that we included test cases under every function. If you run the project_01_template.py file at this point it should print False for each test. After you write the correct code for each function, and then run the file, it should print True for each test. 1. Write a function named gc_content that takes one argument seq and performs the following tasks: a. Counts the number of G and C characters in the sequence and adds these counts. b. Divides the GC count total by the length of the sequence and returns this fraction. 2. Write a function named get_orf that takes one argument seq and performs the following tasks: a. The function assumes that the given DNA sequence begins with a start codon ATG. b. Finds the first in-frame stop codon, and returns the sequence from the start to that stop codon. The sequence that is returned should include the start codon but not the stop codon. c. If there is no in-frame stop codon, get_orf should assume that the reading frame extends through the end of the sequence and simply return the entire sequence. Note: This function is very similar (but not identical) to the one you wrote for Assignment 5. 3. Write a function named one_frame that takes one argument seq and performs the following tasks: a. The function searches given DNA string from left to right in multiples of three nucleotides (in a single reading frame). b. When it hits a start codon ATG it calls get_orf on the slice of the string beginning at that start codon. c. The ORF returned by get_orf is added to a list of ORFs. d. The function skips ahead in the DNA string to the point right after the ORF that we just found and starts looking for the next ORF. e. Steps a through d are repeated until we have traversed the entire DNA string. The function should return a list of all ORFs it has found. Hint: A while loop that uses a call to get_orf will be very convenient here. A function could be written using a for loop, but it may be more difficult. Here is one example of one_frame function in action: >>> one_frame('AATGCCATGTGAATGCCCTAA') ['ATG', 'ATGCCC'1 Note that the first ATG (index [1:4]) was not returned. This is because the first ATG is not in the frame that one_frame is searching. We will search those other frames by additional calls to one_frame in the next steps of this project. Here is another example of one_frame function in action: >>> one_frame('ATGCCCATGGGGAAATTTTGACCC') ['ATGCCCATGGGGAAATTT'] Note that, in this case, there is a second ATG in the sequence (index [6:9]). This ATG is part of an ORF which ends with the same stop codon as the first ORF (that is, it is a smaller nested open reading frame). one_frame skipped this second ORF when it jumped ahead to the end of the first ORF, while looking for a stop codon. This is exactly what we want one_frame to do; we are focusing on large open reading frames, and will skip small nested ones. 4. The next step is to write a function named forward_frames that takes one argument seq. This function will identify all the ORFs in the sequence (without considering the reverse-complement of the sequence), and return them as a list. If there are none, it should return an empty list. Once again, this task is made easier by making use of functions that we have already written: a. First, forward_frames should call one_frame in each of the three possible forward reading frames of the sequence. b. It should then combine all of the ORFs found in each frame and return a list containing these ORFS. Consider how forward_frames should behave given the following sequence: >>> dna = 'CAGCTCCAATGTTTTAACCCCCCCC We can look at the three frames of this sequence by slicing off 0, 1 or 2 base pairs at the beginning: >>> one_frame (dna) 0] >>> one_frame (dna [1:]) 0] >>> one_frame (dna [2:]) ['ATGTTT'] Each call to one_frame will produce a list. You can then combine the lists from the three calls into one list and return it. To combine two lists, you can do the following: >>> list_a= [1, 2] >>> list_b = [3, 4] >>> combined_list = list_a + list_b >>> combined_list [1, 2, 3, 4] Here are a couple examples of forward_frames function in action: >>> forward_frames ("ATGGGATGAATTAACCATGCCCTAA") ['ATGGGA', 'ATGCCC', 'ATGAAT'] >>> forward_frames ("GGAGTAAGGGGG') 5. Finally, write a function named gene_finder that takes three arguments: fasta_file, min_len, min_gc and performs the following tasks: a. Reads a FASTA formatted file fasta file that contains one DNA sequence. You will find a function read_one_seq_fasta in the provided template file: project_01_template.py. Make sure your input FASTA file is in the same directory as your Python file. b. Identifies ORFs longer than min_len (an integer), and GC content larger than min_gc (a float between 0 and 1). c. The function should return a list of lists. Specifically, for each ORF in the sequence that meets the minimum requirements, the function should return a list with the following items: 1. Length of an ORF sequence 2. GC Content of an ORF sequence 3. An ORF sequence itself Below is an example of gene finder function in action. The file gene_finder_test.fasta can be found on Blackboard: >>> gene_finder ('gene_finder_test.fasta', 6, 0.45) [[6, 0.6666666666666666, ATGCCC'], [9, 0.7777777777777778, ATGCCCCGG']]
Step by Step Solution
There are 3 Steps involved in it
Step: 1
a A quick look using less indicates that this is a valid FASTA file I did a quick check to see how many sequences there are by searching for the symbol using grep In2 grep datasalmonellaspi1regionfna ...
Get Instant Access to Expert-Tailored Solutions
See step-by-step solutions with expert insights and AI powered tools for academic success
Step: 2

Step: 3
Ace Your Homework with AI
Get the answers you need in no time with our AI-driven, step-by-step assistance
Get Started
Physics
Authors: Alan Giambattista, Betty Richardson, Robert Richardson
2nd edition
77339681, 978-0077339685