Using the case study listed below, I need help with the following question Which option do you think management should take and provide justification? Think
Using the case study listed below, I need help with the following question
Which option do you think management should take and provide justification? Think about this as you have two stages of options, one for the pilot plant, one for production.
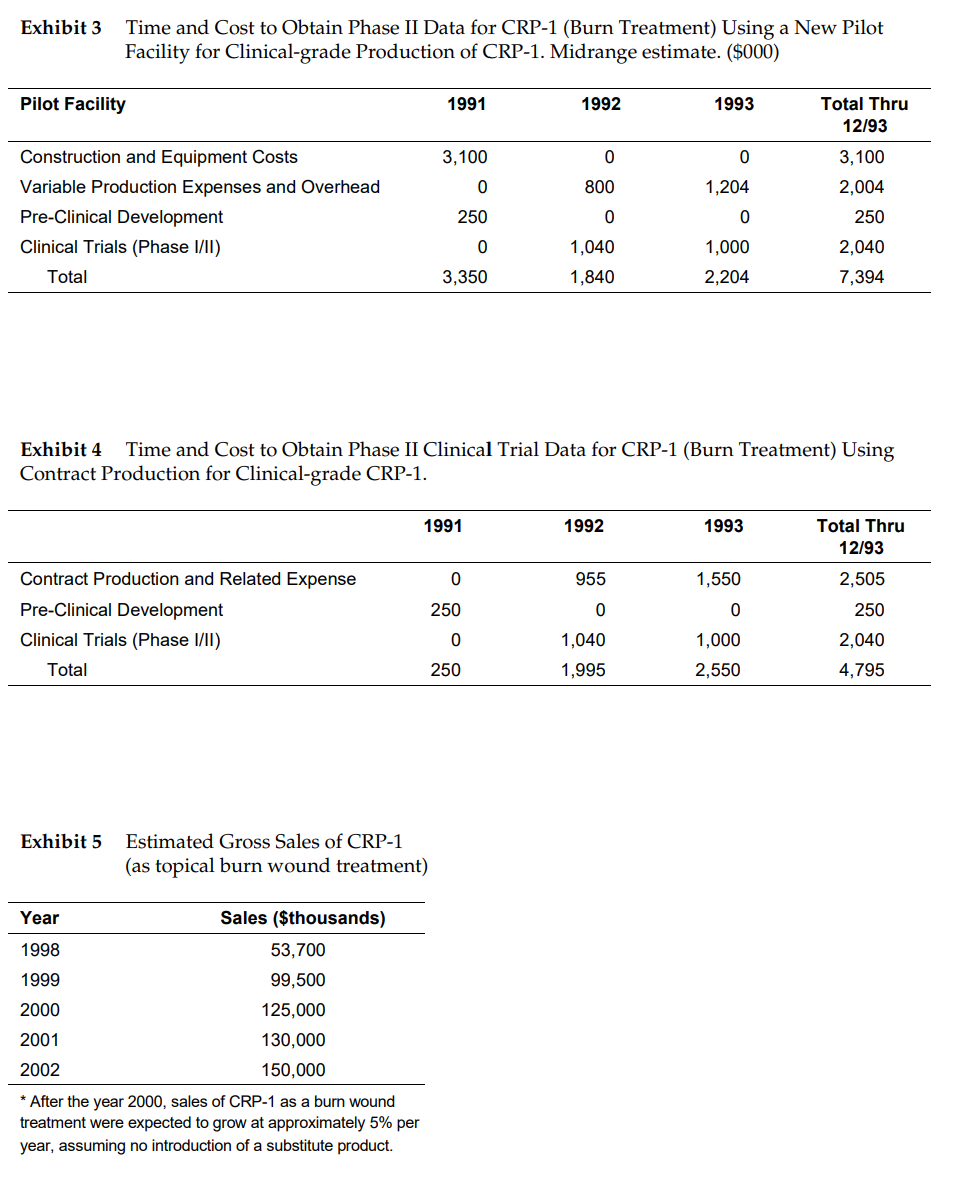
inseam Harvard Business School 9-692-041 Rev. April 14, 1994 Nucleon, Inc. Robert Moore, a recent graduate of a top-ranked MBA. program, now realized what it was like to be on the other side of a case study. It was December 1990 and Nucleon, the young biotechnology start-up at which he had recently become project manager, faced critical manufacturing choices. Moore and Jeff Hurst, the firm's CEO, had met to discuss the situation, and within the next few weeks, Hurst needed to present the company's manufacturing strategy to the board of directors. In the meantime, he asked Moore to evaluate in detail Nucleon's options and give his own recommendation. Nucleon's first potential product, \"cell regulating protein-1\" (CRP-l), had been undergoing extensive experimentation and analysis in the company's R&D laboratories for several years. The next major hurdle was human clinical trials, which also typically took place over several years. However, before Nucleon could launch clinical trials, it had to decide how and where CRP-l would be manufactured. To ensure participants' safety, the US. Food and Drug Administration (FDA) imposed strict guidelines; products being tested in humans had to be made in facilities certified for \"clinical grade\" production.1 Since CRP-l was the company's first product to go into the clinic, Nucleon had no manufacturing facilities which met FDA requirements. It was faced with three options for supplying CRP-l to the clinic: The first was to build a new 5000 square-foot pilot plant with enough capacity to supply all the CRP-i needed for Phases 1 and II of clinical trials. The second option was to contract clinical manufacturing to an outside firm. And a third option was to license the manufacturing to another biotechnology company or to a pharmaceutical firm. Under this third option, the licensee would be responsible for all manufacturing, clinical development, and eventual marketing of CRP-l. Definite risks and rewards were attached to each option, and Moore knew that the one ultimately chosen by Hurst would have long-term consequences for Nucleon's survival in the intensively competitive and high-stakes drug industry. One of contract manufacturing's biggest risks was confidential information disclosure. It was virtually impossible for any contractor to provide reliable time and cost estimates without knowing many proprietary product details. Moreover, the complexity of the products and processes made estimates of time and cost painstaking; reaching an agreement could take many months. Even after a contract was signed, technology transfer and scale-up might take another nine months. Moore noted, \"It will take about as much time to negotiate an agreement and transfer and validate the process as it will for us to build a pilot plant.\" Although production contracts were negotiated typically for fixed quantities (e.g., 100 g of CRP-l over 10 months), in contract negotiations, a balance needed to be struck. On one hand, it was risky to commit to large quantities of material--which might not be required if product specifications changed or the product was pulled from the clinic. On the other hand, short-term contracts usually involved a higher price to offset fixed costs of scale-up and batch set-ups. Under either the pilot plant or the contract manufacturing option, Nucleon would retain ownership of the product rights at least until the commencement of Phase III clinical trials. At that point, the company would enter into licensing and marketing with a large corporate partner. The options for Phase III trials and beyond are discussed later. Licensing the Product to a Another Company Rather than waiting until Phase II] trials to enter a licensing deal, Nucleon could license the product immediately in exchange for fixed payments and future royalties. Under this option, the licensed partner, not Nucleon, would make all the requisite expenditures in clinical development, clinical manufacturing, regulatory filings, and commercial manufacturing and marketing. The partner would have the right to market CRP-l to treat burn wounds. Nucleon would retain the right to develop CRP-l for other therapeutic applications. Nucleon also would receive an up-front, fixed licensing fee and reimbursement for any additional development work it performed on the project. If and when CRP-l was commercialized, Nucleon would receive royalties as a percentage of sales. This licensing option had the chief benefit of generating cash inunediately,' it also spared Nucleon from making large capital investments in clinical development and manufacturing, and allowed the company to concentrate all of its financial and human resources on R&D. Of course, if the product turned out to be successful, Nucleon would receive far lower revenues than if it had made all of these investments itself. Some Nucleon employees viewed this option as "mortgaging away" the company's future. Whether this option would mortgage the company's future depended upon the exact terms of the agreement that Nucleon could negotiate. While it was virtually impossible to know for sure what kind of deal could be struck, Nucleon management had conducted preliminary discussions with several firms. From these and consultations with the company's venture capitalists, Moore determined that Nucleon could expect to reach an agreement with the following terms: Upon signing the contract Nucleon would get a $3 million payment. After the FDA approved CRP-l for burn wounds, Nucleon would receive annual royalty payments from the partner equivalent to 5% of gross sales {Exhibit 5). Manufacturing Options for Phase III and Commercialization One of the Chief advantages of either in-house pilot manufacturing or contract manufacturing over immediate licensing was that it gave Nucleon more options if the project survived Phase I and Phase [I trials. As noted earlier, under these two options, Nucleon intended to line up with a partner who would be responsible for conducting Phase III trials, handling regulatory filings and marketing the product. However, under such an arrangement, Nucleon could either retain commercial manufacturing responsibilities or license these to the partner. Each of these approaches are discussed below. Vertically Integrate into Commercial Manufacturing Before Phase II! trials began, Nucleon could invest in a full-scale commercial manufacturing facility which met the FDA guidelines for Good Manufacturing Practice (Exhibit 6). The FDA required that Phase III trials be supplied largely by the plant which would be used to supply the commercial market. Thus it would be necessary to commence construction in mid-1993 so that the plant could be fully validated and operational by the scheduled commencement of Phase III trials. Moore estimated that the costs of such a facility would be about $20 million, and another $1 million in development resources would be required to perform scale-up. He and Nucleon's nancial advisors believed that once the project cleared Phase II trials, it would have little difficulty raising the needed funds to build the plant. The company would also have to hire at least 20 people to handle such functions as procurement, quality control, maintenance, technical support, and 4 logistics. It was difficult to know exactly what terms could be reached. If Nucleon built a commercial plant, it would be the sole supplier of CRP-l to its marketing partner. Judging by what other firms in the industry were receiving for similar products, Nucleon management estimated that the company Could negotiate a combined supply contract and royalty agreement with the following terms: Nucleon would receive a $5 million payment upon FDA approval of CRP-l plus royalties equal to 40% of the partner's gross sales of the product. Nucleon would sell CRP-l to the partner at cost.5 Licensing Out Manufacturing and Marketing Rights at Phase III A second option at the beginning of Phase Ell trials would be to license out both the manufacturing and marketing rights to a partner. This option would be similar to that discussed above, except, in this case, the partner would also be responsible for Phase III and commercial manufacturing. Nucleon therefore would not have to invest the $20 million in a commercial plant. Under this option, Nucleon could expect to receive a $7 million payment if and when CRP-l was approved by the FDA. After that, Nucleon would receive a royalty equivalent to 10% of the partner's gross sales of CRP-l. Moore recognized that Hurst leaned towards manufacturing CRP-l in-house. He had said, "I keep asking myself, 'How many times will we get to the plate?' If I thought that this were our only chance, I'd go for the home run and take the risks of manufacturing.\" For his part, Moore decided to review Nucleon's options another time before making a recommendation. Exhibit 3 Time and Cost to Obtain Phase II Data for CRP-1 (Burn Treatment) Using a New Pilot Facility for Clinical-grade Production of CRP-1. Midrange estimate. ($000) Pilot Facility 1991 1992 1993 Total Thru 12/93 Construction and Equipment Costs 3, 100 0 0 3, 100 Variable Production Expenses and Overhead 800 1,204 2,004 Pre-Clinical Development 250 0 0 250 Clinical Trials (Phase I/Il) 1,040 1,000 2,040 Total 3,350 1,840 2,204 7,394 Exhibit 4 Time and Cost to Obtain Phase II Clinical Trial Data for CRP-1 (Burn Treatment) Using Contract Production for Clinical-grade CRP-1. 1991 1992 1993 Total Thru 12/93 Contract Production and Related Expense 0 955 1,550 2,505 Pre-Clinical Development 250 250 Clinical Trials (Phase I/Il) 0 1,040 1,000 2,040 Total 250 1,995 2,550 4,795 Exhibit 5 Estimated Gross Sales of CRP-1 (as topical burn wound treatment) Year Sales ($thousands) 1998 53,700 1999 99,500 2000 125,000 2001 130,000 2002 150,000 After the year 2000, sales of CRP-1 as a burn wound treatment were expected to grow at approximately 5% per year, assuming no introduction of a substitute product.Exhibit 1 Approximate Time Frame for CRP-1 Project April 1992 Begin Phase I Clinical Trials December 1992 Begin Phase II Clinical Trials December 1993 End Phase II Clinical Trials June 1994 Begin Phase III Clinical Trials December 1996 Complete Phase III Clinical Trials; File data with FDA January 1998 Expected FDA approval and commencement of salesBackground Nucleon was founded in 1985 by Dr. Alan Ball, an internationally respected researcher at the Children's Hospital and an Associate Professor of Clinical Medicine at the Greaves Medical Center, to develop pharmaceutical products based on a class of proteins known as cell regulating factors. From 1985 to 1988, Dr. Ball and a small group of scientists who joined Nucleon researched ways of producing CRP-l outside the human body. Although CRPl was a naturally occurring protein contained in human blood plasma, the amount that could be extracted was far too small to be of any Commercial use. Scientists first isolated a small amount of naturally occurring CRP-1 and determined the gene that instructed human cells how to produce CRP-l. The gene was then cloned. While this laboratory process for producing CRP-1 was still very small scale, it generated enough material to send to academic collaborators who were exploring the potential therapeutic uses of CRP-1. Although an actual product was still several years and millions of dollars away, early research indicated that CRP- 1 had potential as a treatment for burns and kidney failure. Strategy and Competition Nucleon was one of over 200 firms founded since the mid-19705 to develop pharmaceutical technologies based on recent advances in molecular biology and immunology. This new field of R&D, commonly called \"biotechnology," also attracted the attention of established companies. By 1989, most of the world's largest pharmaceutical enterprises, like Eli Lilly, Merck, and Hoffman LaRoche, had extensive in-house biotechnology R&D programs as well as collaborative ties with many of the new entrants. Competition was intense. Scientists at both start-up and established companies were racing to be the first to clone certain genes and establish proprietary positions for their firms in emerging areas like cell regulating factors. Establishing a strong patent position was particularly important for small companies like Nucleon. Moore explained: "Given the enormous costs of developing and commercializing a new drug, potential investors want to see a strong proprietary position before they commit serious capital. Just one strong patent on the right molecule can ensure survival for years by allowing you to attract capital." Biotechnology patent law, however, was as new and uncertain as the technology itself. Indeed, the legality of patenting a genetically engineered microorganism was only established in 1980 by a landmark United States Supreme Court decision, and the ensuing decade saw many legal battles over the scope and efficacy of specific patents. In some cases, two or more companies had claims on different proprietary elements of the same molecule. For example, one company might claim ownership of the molecule itself while another of the genetic sequence used to synthesize the molecule. Further, it was extremely difficult to patent the process technology used to obtain a biologically important molecule, even though the starting material and the resulting molecule were considered original enough to be patented. Given the lack of precedent, it was always difficult to predict how the Courts would rule in any given situation. Moreover, the United States Patent Office might take several years to process an application. And while few companies could afford to wait until a patent was granted before continuing development, there were big risks in going ahead with development before the granting of a patent. A company could spend tens of millions of dollars in clinical trials and manufacturing facilities yet wind up not having a proprietary position if the patent office denied the application. Even if patents were granted, it was always possible for a competitor to challenge them in court. While Nucleon believed it had a strong patent position on the CRP-l molecule, its rights to other necessary proprietary components {such as the genetic sequence) were less certain. Nucleon management believed that several factors were critical to the company's survival. As Hurst commented: Given how small we are, it's absolutely essential that we pick the right projects. We can't hedge our bets with a big portfolio of projects, like the big pharmaceutical companies can. We've got to pick winners the first time. Gordon Banks, Nucleon's vice president of R&D, and one of the leading scientists in the field of C31] regulating factors added; That's why it's so important for us to be at the leading edge of scientific research. This means not only attracting the best in-house scientists, but also maintaining close contact with universities. If someone at a university clones the genes for a new cell regulating factor, we want to know about it. Nucleon management believed that it had found an attractive niche: relatively few firms were working on cell regulating proteins. Banks believed that the company's distinctive technical capability lay in its ability to identify potentially therapeutic cell regulating factors. Although Nucleon was a leader in cell regulating factors, the company was not free from competition. Other companies were developing drugs using somewhat similar technology. Also, many companies were using alternative technologies to develop drugs for some of the same diseases for which cell regulating factors were being developed. As Hurst commented, \"We're a leader, but we're not alone. It's important for us to get our products into the clinic before others do." Biotechnology firms were using different strategies for developing and commercializing their technologies. Virtually all the biotechnology companies started, like Nucleon, as specialized R&D laboratories. Over time, some vertically integrated into production, and a few of the oldest companies, like Genentech, were even vertically integrating into marketing. Nucleon was presently contemplating its manufacturing strategy. Its marketing strategy, however, was Clear. Nucleon management believed that the company could not afford to market its products on its own. Instead, it planned to link up with established pharmaceutical companies, with strong distribution capabilities, to market its products. Hurst, who once worked in marketing for a large pharmaceutical company, noted: Companies like Merck have hundreds of salespeople. They can reach every doctor's office in the country within one week. It would be crazy for a company like us to go up against them in marketing. Besides, our products are likely to be targeted at a variety of therapeutic markets. We would need a few hundred salespeople to market all these products directly. We're much better off linking up with the best company in each therapeutic market. By December 1990, the privately held company had grown to 22 employees, 18 of whom were engaged in R&D; of these about one-third had PhDs from scientific disciplines such as biochemistry, molecular biology, protein chemistry, and immunology. Most of the R&D staff had been recruited from leading university research laboratories and were strongly attracted to cutting- edge, product-oriented research. Nucleon's size and entrepreneurial spirit created an academic atmosphere in R&D and tight links to the academic/ scientific community. Since its founding, Nucleon had raised approximately $6 million in venture capital and received research grants from the US. Department of Agriculture totaling $600,000. Drug Development: From Research to Market Establishing the safety and efficacy of products like CRP-l that were based upon novel genetic engineering technology was enormously complex, time-consuming, and expensive. Nucleon's drug development process, divided into several distinct phases, is discussed below. Research Before launching a research project to develop a new drug, Nucleon management considered several factors in evaluating a project's profit potential. First, there had to be a chance of achieving a dominant proprietary position. Second, the market had to be large enough to justify the R&D investment. Finally, Nucleon wanted to develop drugs where no alternative treatments were available. During the research phase, Nucleon's Scientists sought to identify and purify from human plasma minute quantities of cell regulating proteins that might have therapeutic value. Some critical information to pursue this research was obtained by perusing scientific literature or by consulting with leading academic researchers. Much necessary information, however, was still undiscovered and came only from in-house research and experimentation, which seldom moved in a straight- forward, logical manner but from one obstacle to the next. This could entail abandoning one strategy and starting over again. Cloning and Purification Products like CRP-l and others that Nucleon intended to develop were fundamentally different from most drugs developed by pharmaceutical companies, which traditionally were synthetic chemicals. Chemical synthesis was effective for relatively small and simple molecules, but proteins like CRP-l were simply too big and complex to be synthesized that way and instead were produced by genetic engineering. Through genetic engineering, the scientist created a microscopic protein factory. The gene for the protein was identified, isolated, and cloned, then inserted into different strains of the bacterium E. coli. In theory, the genetically engineered bacteria could then produce the protein in a test tube or shake ask. However, since genetic engineering was still a relatively new scientific discipline, it was not always easy to either identify the relevant genes or to get \"host\" (genetically altered) bacteria to produce a specific protein. In practice, it was usually necessary to try different types of host cells to find one or more capable of producing the protein in quantities that could be scaled-up to an economically feasible process. Only a few milligrams of protein could be produced from genetically engineered cells grown in shake asks. Thus, an extensive amount of work then had to go into developing the processes for making each of these proteins in large quantity. Pre-Clinical Research Before a pharmaceutical was tested in humans it underwent pre-clinical evaluation, consisting of experiments in animals to evaluate its efficacy. Over six to eight months, increasing doses were administered to animals with and without the simulated disease. Another six months might be needed to evaluate the data. By this point, the company might have spent $6 to $10 million in R&D and preparation of regulatory documents. Only after completing all the requisite animal tests, and having a suitable production process, could the company file for permission with the FDA to commence clinical trials in humans. Though Nucleon had not begun human clinical trials, management expected to file an application with the FDA to begin human trials for CRP-l as a burn wound treatment in 1992. The company was also doing research to determine if CRP-l might have other therapeutic applications. There was some preliminary data suggesting that it might treat kidney failure. Moore estimated that about another two years and $3 million of work were needed before the kidney failure application could be tested in the clinic. Human Clinical Trials Most governments required every new pharmaceutical product to undergo extensive clinical testing before it could be marketed widely, and the FDA regulations were considered the most stringent in the world. To meet them, any new drug, or any approved drug being modified for a different therapeutic application, had to undergo three phases of clinical trials. Phase I trials assessed basic safety. During these trials, the drug was administered to a small group of healthy volunteers and any adverse reactions (such as fevers, dizziness, or nausea) were noted.2 This phase usually required between 6 and 12 months. As long as there were no serious side effects, the product moved to Phase II trials where it was administered to a small group of patients having the disease the drug was presumed to treat. The patients were monitored to determine whether their condition improved as a result of the drug and whether they suffered any adverse side effects. It was during Phase II trials that appropriate dosages were determined. This phase typically required between one to two years to complete. If Phase [I trials succeeded, the product then moved to Phase III trials. Phase III trials assessed the product's efficacy with a relatively large sample of patients on a statistically rigorous basis. Typically, these trials involved multiple hospitals and could require from two to five years to complete. Because of the large number of patients, doctors, and hospitals involved, this stage was by far the most expensive. The costs of manufacturing the drug, administering it to patients, monitoring results, analyzing data, and preparing the requisite regulatory paperwork could run between $530-$100 million. It was imperative for regulatory reasons to manufacture the product with the same process that would be used when the product was marketed commercially. Any change in manufacturing would mean repeating human clinical trials to prove that the deviation did not alter the product's safety and efficacy. This also added significantly to the costs of running Phase III clinical trials. The CRP-1 Project: Current Applications Since Nucleon's founding, its main development project had been CRP-l and most of the company's R&D resources had been focused on the CRP-l projects. While CRP-l's commercialization was still a few years away, Nucleon's scientists and investors were optimistic about its potential. Exhibit 1 depicts the expected time to FDA approval. Initial research focused on developing two major therapeutic applicationsone for topical treatment of burn wounds, the other for acute kidney failure. Both the burn wound and kidney failure markets were estimated to be similar in size. Furthermore, in 1988, the company had also begun investigating two new cell regulating factors, still in the early stages of research. Dr. Banks estimated that these could be ready for clinical trials in about four years if the company spent $10 million on each one. One of the most critical activities currently taking place on the CRP-l project was the development of a larger scale production process, with sufficient capacity to meet all clinical trial requirements. Every step of the process had to be carefully documented and validated to ensure that it could produce identical product from batch to batch. Process Development and Manufacturing CRP-l production would require four basic process steps: 1) fermentation, 2} purification, 3) formulation, 4) filling and packaging (see Exhibit 2). Fermentation Fermentation initially focused on growing the genetically engineered E. cuff in small laboratory asks; the process was then scaled up to successively larger vessels. Unfortunately, the process used to grow cells in a 1-liter glass bottle might not work when attempted in a 10-liter glass chamber or a lU-liter stainless steel tank (also known as a fermentor or bioreactor), given differences in heat exchange, tank aeration, and fermentor geometry. The kinds of nutrients cells were fed, bioreactor temperature, acidity level, oxygen ow rate into the bioreactor, and dozens of other process parameters, were all determined during fermentation process development. While crude fermentation processes existed for over 6000 years, fermentation using genetically engineered cells dated to the early 19805. Many biotechnology firms encountered major difficulties when trying to run pilot and commercial scale fermentation processes for the first time, as Dr. Ann Dawson, Nucleon's director of process science, explained: There are so many unknowns and so many things which can go wrong. If a virus gets into your bioreactor, you could be shut down for weeks. Incredibly tight process control is an absolute must, and even then, you may still run into troubles. For regulatory reasons, it was absolutely critical to run the process exactly as specified. Such strict adherence to process specifications was necessary because even minor process deviations could impact product quality. In addition, the efficiency of the process could be severely affected by Changes in any one of the key process parameters. Because such production methods were new, process development required a great deal of trial-and-error and close collaboration between research scientists and process development scientists early on in the project. Research scientists had to design a process that worked in a test tube as well as on a larger scale, and process development scientists had to be aware of and understand the details of the product and its host cell. Some genetically-engineered cell lines, for example, were extremely difficult to grow large-scale. Dr. Dawson noted: Ideally, we want the research scientists to work with only those cell lines which we know can be scaled-up. While I think they agree with this in principle, they really don't want to be constrained, particularly if they're having trouble getting expression with one of our \"preferred\" cell lines. While much progress had been made over the past decade, many people corlsidered biotechnology production processes very much an art. It was not unheard of for a process to work well in one facility but fail completely when transferred to another. One Nucleon researcher who had experience with such transfers explained: You would be surprised at all the little things that could be done differently from one organization to another. Most of these things are so minor you would not even think of writing them down. But they make the difference between a successful and unsuccessful process. Currently, Nucleon had scaled up the process for making CRP-l to 1!] liters, enough to Supply material for its own biochemical studies, academic collaborators, and potential joint venture partners who wanted to evaluate the product. Early phase clinical trials were likely to require a 100- liter process, and commercial production a much larger scale process. As complex as it was, bacterial fermentation was considered one of the more efficient ways of producing proteins like CRP-l. in some cases, product characteristics could be enhanced if a mammalian cell (e.g., from a mouse or human) rather than a bacterial cell was used as a host. Mammalian cell processes, while desirable from the product side, were much more complex than bacterial processes, not well understood, and much more expensive to maintain. Mammalian cells had to be fed more expensive nutrients, and they grew much more slowly than bacterial cells. They required different bioreactors, and even stricter adherence to original process specifications than bacterial cells. Dr. Dawson explained, "With bacterial cells, a one degree Centigrade temperature change can slow the growth rate and increase your costs, whereas mammalian cells might just die altogether. " Although most biotechnology companies had some experience with bacterial cell processes, many fewer had mammalian cell capabilities. Fortunately, CRP-l could be produced using bacterial cells. The company's R&D lab, however, was already working on second generation CRPl molecules produced in mammalian cells. And biotechnology companies overall were unsure whether existing process development technology would be viable in the future to produce biological products.3 Purication After fermentation, the cells would be broken apart and the CRP-l protein separated from all other proteins and cell debris contained in the fermentation tank. A series of fractionation and centrifugation steps would isolate the cell protein from carbohydrates, fatty acids, and DNA. The CRP-l containing protein mixture would then be purified in three additional steps using a filtration procedure known as column chromatography. Like fermentation, the purification process specified during process development had to be strictly followed during manufacturing. After purification, the material would be subjected to extensive quality testing to ensure that the product met the FDA's extremely high purity standards. Formulation Purification processes yielded nearly pure quantities of the protein of interest, for example CRP-l. At this stage, the product was made into the intended dosage form (e.g., oral, topical, injectable), and subjected to extensive quality testing. For burn treatment, CRP-l would be formulated into a topical dosage form. Filling and Packaging During the final step, bulk quantities of the formulated product were put into tubes, bottles, or other vessels required for administration to patients. The sealed vessels were then inserted into packages, which were also sealed. The Financial Environment A critical issue affecting Nucleon was capital availability. The situation had changed dramatically since the late 19705 and early 1980s when investors lined up to provide capital to brand new biotechnology companies. By the mid- to late-19805, private and public equity markets grew tighter, and venture capitalists, who expected investment returns of 30%, became more selective. The 3011 the horizon was a hybrid of biotechnology and synthetic chemical techniques that could alter or replace existing process technologies. These hybrid companies used molecular biology techniques to clone and produce small amounts of biologically important proteins. The protein was studied to learn the chemical and physical structure of its therapeutically active site and then, using computeraided modelling techniques, the active site of the protein could be constructed synthetically. state of the public equity markets in 1990 made "going public\" virtually impossible for a company like Nucleon; furthermore, potential corporate partners, who had been disappointed by previous biotechnology relationships, were unwilling to fund early stage projects. As Hurst described it: In the early 1980s, a company like ours could have gotten corporate funding with just our idea. By the mid-1980s, we probably would have needed to have started some lab work and have had some preliminary experimental data. Now, it's hard to get a large pharmaceutical company to talk to you unless you've got some solid Phase I and Phase II clinical results and can demonstrate that you've got a stable manufacturing process. And even then, they'll cut some pretty tough terms with you. When it comes to raising capital today, it's a buyers' market. Nucleon was just about to receive another $6.0 million infusion from its venture capitalist. This funding, combined with existing cash on-hand, revenues, interest, and grants, would give Nucleon about $6.5 million. Furthermore, if CRP-l showed promise in pre-clinical trials, Hurst felt that Nucleon could raise enough money to pursue Phase I and II clinical trials. Some analysts were predicting that by 1991 or 1992, Wall Street would once again find biotechnology stocks attractive and there would be opportunities for smaller companies to raise money by sellng stock to the public. Others thought the capital situation would stay tight for at least several more years. The possibility that a long awaited \"shake-out\" was about to hit the biotechnology industry was making many investors cautious. One promising sign was that large corporations again seemed willing to fund some selected projects at very early stages of research. As Moore noted, \"Today, some brand new start-ups in new fields, like antisense, are cutting some deals on projects which are still years away from the clinic." Manufacturing Options for Phase I and Phase II CRP-1 Development Nucleon management contemplated three options to produce clinical grade CRP-l: 1} build a new pilot facility, 2) contract CRPl production to a third-party, or 3) license manufacturing and marketing rights to another biotechnology company or pharmaceutical firm in exchange for up front cash payments and royalties on future product sales. Each option is described below. The New Pilot Plant Nucleon commissioned an engineering consulting firm to study the physical requirements and costs of a new pilot plant (Exhibit 3). The proposed 5000 square-foot plant would be fully equipped with all the state-ofthe-art processing equipment and environmental controls necessary to meet clinical production standards. Planned capacity would meet Nucleon's requirements for Phase t and Phase II clinical trials. The pilot facility, however, could not be used to produce CRP-l for Phase III trials, because it would not meet FDA manufacturing standards for those trials. It was beyond Nucleon's financial capability to build such a plant at this time. The main advantage in building a pilot plant, as Moore saw it, was that it would enable the firm to develop the nucleus of a future larger-scale, in-house manufacturing capability. Because most of Nuclenn's employees were PhD scientists engaged in R&D, it currently lacked supervisors and technicians who could carry out the maintenance, procurement, quality assurance, technical support, logistics, and other functions to operate even a small manufacturing plant. Recruiting people with the appropriate skills and getting the manufacturing organization to work effectively would take time. Supplying clinical trials would allow manufacturing time to accumulate experience dealing with many complicated technical and regulatory issues. Moore noted: If Nucleon waits until Phase EII trials to bring manufacturing in-house, we might find ourselves with a "green" manufacturing organization just when the stakes are highest. By starting now, we'll have the basic manufacturing skills in- house and ready to go when we are really going to need them. The second big advantage of the pilot facility is that it would keep control over process and quality procedures firmly in Nucleon's hands. Dawson added, "Scaling up will be much easier if we have our own pilot plant to experiment in.\" Of course, building a pilot plant was risky. Moore knew that despite its promise in laboratory experiments, it was uncertain at this point how well CRP-l would work when tested in humans. Indeed, if the history of the pharmaceutical industry was any guide, most drugs that entered clinical trials never reached the market. This high risk of failure was offset somewhat by the fact that CRP-l had several potential therapeutic applications. If clinical trials for burn wounds were not promising, it might be used in other applications. Nevertheless, Nucleon management had to consider the possibility of the pilot plant being idled if CRP-l performed poorly in the clinic. Other products under development were still years away from requiring pilot manufacturing capabilities. Another major risk involved process uncertainty. The pilot plant would be designed to produce products using bacterial fermentation; but the company was already in the early stages of developing a version in mammalian cells, which would require vastly different process development capabilities. Some board members believed that Nucleon should focus all of its financial, managerial, and technical resources on R&D. Manufacturing, they felt, would only distract the company from its main mission of exploiting its unique scientific capabilities in the discovery of cell regulating proteins. According to Hurst: Our venture capitalists are asking us where we, as a company, add the most value. As a small research-intensive company, we can be the \"fastest guns on the block" when it comes to drug discovery. But that means funneling our limited resources into R&D. Some of our investors are concerned that we could get bogged down in manufacturing. On the other hand, it's getting to the point where anyone can clone a gene. I keep wondering whether we can still differentiate ourselves on R&D alone. Contract Manufacturing Contracting manufacturing was a second option for Phase I and Phase II CRP-l development. The biggest advantage of this option was that it required no major capital investments on Nucleon's part. If CRP-l failed, the contract could be easily terminated. Aside from relatively Small termination penalties, the company would have little else at risk. Another advantage was that companies supplying contract manufacturing services had facilities and personnel in place. Contract production was not inexpensive (see Exhibit 4). There were very few U.S. companies capable and willing to contract manufacture pharmaceuticals from bacteria. Nucleon management was meeting with several potential contractors. These included other biotechnology companies who had excess capacity. In recent years, many biotechnology companies had built GMI' plants in anticipation of future products. When product approvals were delayed or even rejected by the FDA, these companies found themselves with tremendous excess manufacturing capacity. Because of mounting financial pressures, some of these Companies were providing contract manufacturing services. Some industry experts believed that excess manufacturing capacity would continue to accumulate during the next few years
Step by Step Solution
There are 3 Steps involved in it
Step: 1
Get Instant Access to Expert-Tailored Solutions
See step-by-step solutions with expert insights and AI powered tools for academic success
Step: 2
Step: 3
Ace Your Homework with AI
Get the answers you need in no time with our AI-driven, step-by-step assistance