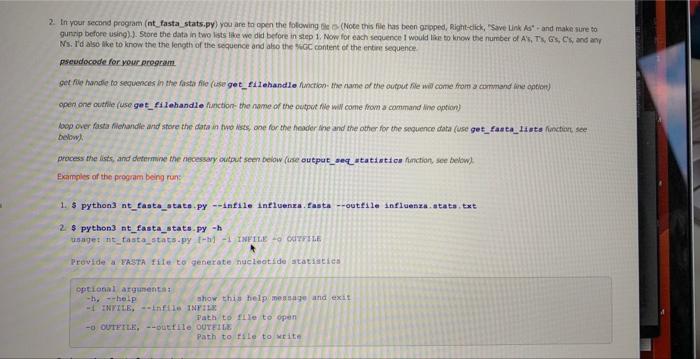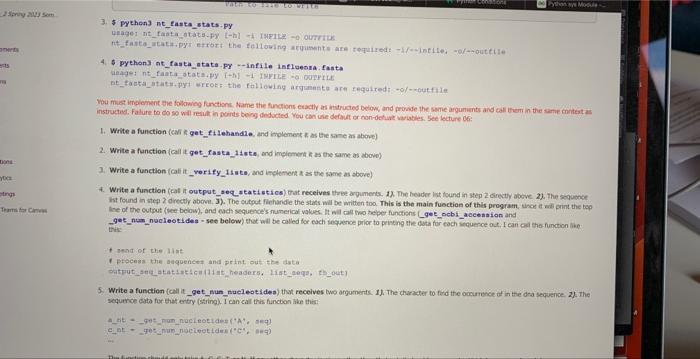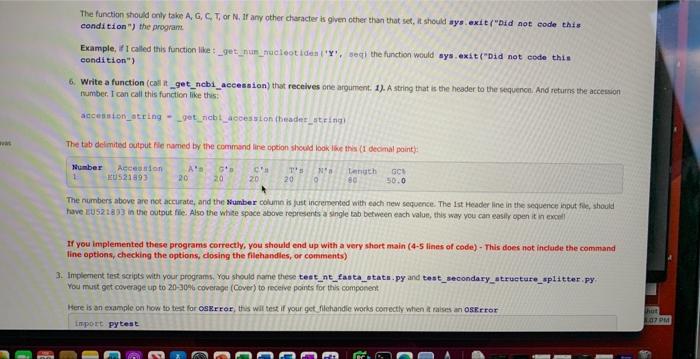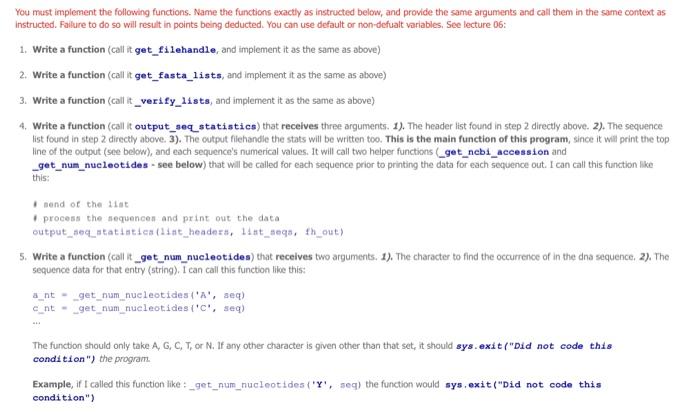

You must implement the following functions. Name the functions exactly as instructed below, and provide the same arguments and call them in the same context as instructed. Failure to do so will result in points being deducted. You can use default or non-defualt variables, See lecture 06: 1. Write a function (call it get filehandle, and implement it as the same as above) 2. Write a function (call it get_fasta_lists, and implement it as the same as above) 3. Write a function (call it _verify_1ists, and implement it as the same as above) 4. Write a function (call it output_seq _statistics) that recelves three arguments. 1). The header list found in step 2 directly above. 2). The sequence list found in step 2 directly above. 3). The output filehandle the stats will be written too. This is the main function of this program, since it will print the top line of the output (see below), and esch sequence's numerical values. It will call two helper functions (_get__nebi_aceession and _get_num_nueleotides - see below) that will be called for each sequence prior to printing the data for each soquence out. I can call this function like this: 9 bend of the diat 1 process the sequencen and print out the data output_seq_atatiatics (1itt_headera, 1itht_segs, ih_out) 5. Write a function (call it _get_num_nucleotides) that recelves two arguments. 1). The character to find the occurrence of in the dna sequence, 2), The sequence data for that entry (string). I can call this function like this: ant= get_num_nueleotides (A, aeq) cnt=get num_nucleotides (C, seq) The function should only take A,G,C,T, or N. If any other character is given other than that set, it should sys. exit ("Did not code this condition ") the program. Example, if I called this function like:_get__ _um_nucleotides (' Y ', seg) the function would sys, exit("Did not code this condition") 1. Write a function (call it get filehandle) that receives two arguments: 1). A file name 2), How to open a file for reading or writing ("r", or "w") in Python. The purpose of this function is to open the file name passed in, and passes back a a file object, aka the file handie or handle. You can call this function like this: \[ \begin{array}{l} \text { fh_in = get_filehandle }\left\{f i l e \_t o \_o p e n, " r * ight) \text { or } \\ \text { fh_out = get_filehandle(file_to_write, "w") } \end{array} \] When using open (), make sure to use try .. except .. except if the open was not successful the program should raise the right Exception(it should raise an oskmor si for when the file cannot be opened, and raise a valueEror se. when the wrong argument was passed for the opening mode, e.g. "mr" instead of " r "). We will test for things like a file that does not exist for opening, or the wrong open mode, e.g. mode='m'. See poan B for more information. All opening and closing of files in your program should use the get_filehandle function. Failure to do so will loose points. Make sure to dose your file hancle. 2. Write a function (call it get_fasta_1ists) that receives one argument: 1). A file handle to the fasta file used in this program. The function will return two lists. One lists for the sequences in the file and one list for the headers to the sequences in the file. There should be a one-to-one correspondence to the data in the lists. Meaning element 1 of the header lst should correspond to element 1 of the sequence list. If implemented correctly, you can call this function like this: I send off the filehandle I get back data in 1ints 1ist headers, Iist_seqs = get_fasta_1ists (fh_in) This function should exit if it could not successfully get two lsts of equal size (see _verify_ 1i sts below). The function _verify_ 1i s ts does the actual exiting and printing of the message, but that will get called to by the get_fasta_1ists function. This function can be tested by a unit test. Make sure "newline n7 characters have been removed from your sequence data, Note, remove newline characters, not spaces, since the secondary structure string might contain spacest 3. Write a function (call it _verify_1ists [Note the " _" in front of the name of the function]) that receives two arguments: 1). The header list found in step 2 directly above. 2). The sequence list found in step 2 directly above. This is a helper function that will be called in the get f fasta 11 ats function (which is why it starts with a " " in front of the name, since it's not really to be called in the main part of your program, Le the Single Pre Underscore is only meant to use for the internal use). If the sizes of the lists passed into this function are not the same, it should exit, (telling the user wiry it exited - see output 6. Write a function (call it _get_ncbi_accession) that recelves one argument. 1). A string that is the header to the sequence. And returns the accession number, I can call this function like this: accession_atring = _get_ncbi_accesaion (header_string) The tab delimited cutput file named by the command line option should look like this (1 decimal point): The numbers above are not accurate, and the Number column is just incremented with each new soguence, The Ist Header line in the sequence input file, should have EU 521893 in the output file. Also the white space above represents a single tab between each value, this way you can easily open it in excell gursip before using).]. Store the data in two lists like we dia before in step 1. Now for each sequence I would lien to know the nurnber of A's, Tis, Gss, Cif and ary Nis, I'd also Mke to know the the length of the sequence and also the oscic content of the entire sequence pseudocode for your Reregrim. opetr one outhie (use get fi lehandle finction- the nwne of the outout fie wil come from a command ane option) bog oier fasta filehandle and store the dota in two ksts, one for the finader the and the other for the snguence data (use get fase ca 14 ats finction, see belinys. process the ists, and deternine the aecessary oulput secen below (cse output_seq_at atist i co functiort, see belbw). Examples of the proeram being rune 1. \$ python3 int fasta stata.PY minfile influenxa. fasta -outfile influenxa. atata.txt 2. \$ python3 nt_fasta_stats-py h Provide a FASTh fite to generate nucleatide statiseich eptional aryunenta a -h. ahelp ahor thig tielpineagage and exit -i HNXILE, =hft1e1NPiLg. Path to 11 re to open a otyterth, --outile oUtediz path to file to write. 3. \$ Pythen3 nt_Easta_stats-Py qiage nt__aita notats-fy {h} of IMPILe 0 ourritus. 4. 5 python3 nt. fasta atata-Fy *infile intleenra. fasta 1. Write a function (caf \& get fi lehandfie, and inplement a as the sane m above) 2. Write a fanction (call it get_fasta 11 tes, and imeioment it as the same as abovo) 3. Write a function (cali it _ verify 1i i t, , and imelement it as the some as above) 4. Write a function (cal it outpat a eq atati stics) trat receives ifrec arpuments. 1.x. The header Int, found in step 2 cirectiv abeve. 2 , The sequence i nend of the list: 1 pipeess the adqoences and print out the dista 5. Write a function (call it_get__Bun_nueleatides) that recelves bwo argurnerts: 1). The charater to fricd the ousirence of in the dra sequence 21. the sequrnce data for that entry (sting). 1 cancall this fincbon hke thert: The function should only take A,G,C,T, or N. If any other character is given other than that set, it should ays.exit ("Did not eode this condi tion") the program Example, if 1 called this function like __get_nun_nieleot tdein ('Y', seql the function would ays.exit ("Did not eode this condition") 6. Write a function (cail it get_ncb1_aceession) that receives one argunsent. 1). A string that is the header to the sequenon. And returns the atcension number, I can call this function like this: accesaien_atring - _get_ncbl_aceesston (header_string) The tab deimited output fic numed by the command lire option should bok lase this (1 deomal point): The numbers above are not accurate, and the Sumber column is lust incremented with eoch new sequense. The ist Header line in the sequence input foe, should If you implemented these programs correctly, you should end up with a very short main (4-5 lines of code) - This does not indude the command line options, checking the options, dosing the filehandies, or comments) Inplement test saripts with your programs. You should name these teet_nt_fasta__tats.py and test_secondary_atructure_splitter. py. You must got coverage up to 2030% covenage (Cover) to receive points for this component Here is an example on how to test for osgrror, ths wil test if your get filchandie works correctly when at raises an oserror