

Question: Molecular orbitals are most commonly delocalized throughout the molecule and exhibit distinct bonding or anti-bonding character. Loss of an electron from a specific molecular orbital
a. Obtain equilibrium geometries for ethene, formaldimine, and formaldehyde using the HF/6-31G* model and display the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) for each. What would happen to the geometry around carbon (remain planar versus pyramidalize), to the C‰¡X bond length, and (for formaldimine) to the C‰¡NH bond angle if an electron were to be removed from the HOMO of ethene, formaldimine, and formaldehyde?
b. Obtain equilibrium geometries for radical cations of ethene, formaldimine, and formaldehyde using the HF/6-31G* model. Are the calculated geometries of these species, in which an electron has been removed from the corresponding neutral molecule, in line with your predictions based on the shape and nodal structure of the HOMO?
Unoccupied molecular orbitals are also delocalized and also show distinct bonding or antibonding character. Normally, this is of no consequence. However, were these orbitals to become occupied (from excitation or from capture of an electron), then changes in molecular geometry would also be expected. What would happen around carbon, to the C‰¡X bond length, and (for formaldimine) to the C‰¡NH bond angle, if an electron were to be added to the LUMO of ethene, formaldimine, and formaldehyde?
c. Obtain equilibrium geometries for the radical anions of ethene, formaldimine, and formaldehyde using the HF/6-31G* model. Are the calculated geometries of these species, in which an electron has been added to the corresponding neutral molecule, in line with your predictions based on the shape and nodal structure of LUMO?
The LUMO is quite successful in anticipating detailed changes caused by electron addition. The first excited state of formaldehyde (the so-called n †’ Ï€* state) can be thought of as arising from the promotion of one electron from the HOMO (in the ground state of formaldehyde) to the LUMO. The experimental equilibrium geometry of the molecule shows lengthening of the C~O bond and a pyramidal carbon (ground-state values are shown in parentheses):
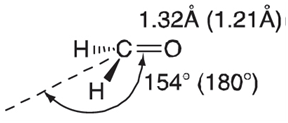
This structure is in qualitative agreement with the results of calculations on the radical cation and radical anion (relative to that on the neutral). Both the radical cation and radical anion show C~O bond lengthening (relative to the neutral molecule). The radical anion shows pyramidalization of the CH2 group. Both features are found in the first excited state of formaldehyde.
d. Rationalize this experimental result on the basis of what you know about the HOMO and LUMO in formaldehyde and your experience with calculations on the radical cation and radical anion of formaldehyde.
1.32 (1.21) H C=0 154 (180)
Step by Step Solution
3.38 Rating (170 Votes )
There are 3 Steps involved in it
a Ethene formaldimine formaldehyde Based on the shape and nodal structure of the HOMO the geometry o... View full answer

Get step-by-step solutions from verified subject matter experts